Target Team : Efficacy and Resistence to anti-tumor targeted therapies, David Tulasne
Emergent Team : Molecular Therapeutic Approaches for Genetic Diseases and Cancer, Fabrice Lejeune https://nonsense.univ-lille.fr/
Presentation : Target team
Targeted therapies are currently transforming cancer management and improving life expectancy. Their effectiveness relies on oncogenic addiction, where cancer cells survival and/or growth depend on a single oncogene, often tyrosine kinase receptors (RTKs). Among these, MET is a promising target, with tyrosine kinase inhibitors (TKIs) approved for the treatment of lung cancer. However, their efficacy is limited compared to other targeted therapies.
Our goal is to improve therapies targeting MET through three main areas:
- understanding the mechanisms of oncogenic addiction
- identifying resistance mechanisms
- expanding patient eligibility for targeted therapies.
To achieve this, our team focuses on lung and prostate cancers, the main causes of cancer mortality. Our research provides fundamental knowledge on oncogenic addiction and proposes new therapeutic strategies.
We have deliberately chosen to develop translational research and to work closely with molecular biologists, pathologists and clinicians at the CHU, who are directly involved in patient care.
We are constantly looking for Master students, PhD students, post-doc fellows or colleagues who would be interested in joining us to participate in the development of our research projects.
 | David TULASNE – Research Director (DR2) INSERM Research: Involvement of the MET receptor tyrosine kinase in cancers. Keywords: Receptor tyrosine kinase, Intracellular signaling, Tumor suppressor genes, Survival/apoptosis balance, Transcriptional regulation, lung Cancer, Targeted therapies, Resistance. david.tulasne@cnrs.fr |
 | Martine DUTERQUE-COQUILLAUD – Research Director (DR2) CNRS Research: ETS fusions in prostate cancer and metastasis formation. Keywords: Prostate cancer, Bone metastasis, ETS fusion, Transcriptional regulation. martine.duterque@cnrs.fr |
 | Anne CHOTTEAU-LELIEVRE – Professor (PR2) University of Lille Research: Cooperation between MET receptor and ETS fusions in prostate cancer progression Teaching: Molecular Biology, Genetic, Cellular Biology. Teaching responsibilities: Headmaster of Biotechnologies Master – Headmaster of “Cursus Master Ingénierie « Biotechnologies-Bioingénierie”. Keywords: Cancer, Prostate, ETS transcription factors, MET Receptor, Transcriptionnal regulation. anne.chotteau@univ-lille.fr |
 | Zoulika KHERROUCHE – Researcher (CR1) Institut Pasteur de Lille Research: Mechanisms of resistance to targeted therapies and innovative therapeutic approaches. Keywords: Receptor tyrosine kinase, Intracellular signaling, tumor suppressor genes, Survival/apoptosis balance, Transcriptional regulation, lung Cancer, Targeted therapies, Resistance. zoulika.kherrouche@cnrs.fr |
 | Dr Marie José TRUONG, Researcher, (CR1) Institut Pasteur de Lille Teaching: Creation and supervision of the “Scientific Teachings” department at Pasteur Institute of Lille. Accreditation of Education and Training Courses in Laboratory Animal Science. marie-jose.truong@cnrs.fr |
 | Fabrice LEJEUNE – Researcher (DR2) INSERM https://nonsense.univ-lille.fr/ Research: Correction of nonsense mutations and study of the mechanisms leading to the recognition of premature termination codons. fabrice.lejeune@inserm.fr |
 | Edith BONNELYE – Researcher DR2 CNRS |
| Marie-Hélène VIEILLARD – Onco-Rhumatologist (PH) COL and CHU Lille |
 | Sarah HUMEZ – Anatomo-Pathologist (PH) CHU Lille |
 | Arnauld VILLERS – Urologist – Professor (PU-PH) CHU Lille Research: Translational research in prostate cancer. Risk factors for progression. Metastasogenesis. arnauld.villers@chu-lille.fr |
 | Xavier LEROY – Anatomo-Pathologist – Professor (PU-PH) CHU Lille |
 | Alexis CORTOT – Pneumologist – Professor (PU-PH) CHU Lille |
 | Simon BALDACCI – Pneumologue (MCU-PH) – CHU Lille |
 | Jonathan OLIVIER – MUC-PH-MD-PhD, Urologist CHU Lille |
 | Marie FERNANDES – PhD student University of Lille Involvement of MET alterations in lung cancer. marie.fernandes@cnrs.fr |
 | Catherine AMPEN-GUFFROY – Engineer (IE) CNRS Molecular and Cellular Biology, Biochemistry, Bioinformatics, Animal experimentation, Health and safety (radiation protection advisor) – catherine.leroy@cnrs.fr |
 | Gaëlle REMY – Ingénieure CNRS Chargée de projets réglementaires des expérimentations in vivo. |
 | Nathalie VANPOUILLE – Technician (TL4) Institut Pasteur de Lille |
 | Isabelle DAMOUR – Technician (TL4) Institut Pasteur de Lille |
 | Audrey VINCHENT – Technician (TL2) Institut Pasteur de Lille |
 | Edwina GBAGUIDI-MICOURS – IE INSERM – CCD |
| Lucie GEOFFROY, IE INSERM |

STUDENTS
 | Rémy TELLIER, PhD student, Univ. Lille |
Thomas AMOUYEL, MD, PhD Student Univ. Lille | |
Denis SEGUIER, MD, PhD student Univ. Lille | |
 | Maram ROUIS, PhD student Univ. Lille |
 | Léa BOUVIGNIES, PhD student Univ. Lille |
 | Camille ORANGE, Master2 Recherche, Univ. Lille |
Gautier MARCQ – MCU-PH – MD – Urologist
|
FORMER MEMBERS
 | Anthony TURPIN – MD-PhD, Oncologist CHU Lille Medical Oncologist – Urologic and GI cancers – anthony.turpin@chu-lille.fr |
 | Aline GHESQUIER – PhD Univ. Lille Cancer de la prostate et Covid-19 : rôle des androgènes dans la régulation de l’expression de TMPRSS2, protéase majeure dans l’infection virale au SARS-CoV-2 |
 | Anne-Claire FLOURENS – Technician (TCE) INSERM Molecular biology, Biochemistry, Histology – anne.flourens@cnrs.fr |
 | Martine PALMA – PhD Univ. Lille |
 | Julie CARRARD – Postdoc |
 | Célia GUERIN – PhD Univ. Lille Etude fonctionnelle des nouvelles mutations du récepteur MET dans le cancer rénal héréditaire. |
 | Elisa CAROUGE – PhD Univ. Lille |
 | Pauline PARENT – MD – CHU – University of Lille |
 | Angela MORABITO – Engineer (IE) CDD CNRS Cellular biology, Molecular biology, Animal studies, Histology. angela.morabito@cnrs.fr |
 | Louise KALMUK – Master2 University of Lille MET mutations as resistance to EGFR inhibitors in lung cancer – louise.kalmuk@cnrs.fr |
 | Philippe JAMME – MD, PhD Univ. Lille Etude des mécanismes de résistance aux inhibiteurs tyrosine kinase dans les cancers pulmonaires porteurs d’une addiction oncogénique. |
 | Sonia PAGET – Engineer (IE) CCD CNRS Biochemistry, Molecular biology, Cell culture and Incucyte (cell proliferation, cell migration, wound healing and spheroids). sonia.paget@cnrs.fr |
Targeted therapies are currently transforming cancer management and improving life expectancy. Their effectiveness relies on oncogenic addiction, where cancer cells survival and/or growth depend on a single oncogene, often tyrosine kinase receptors (RTKs). Among these, MET is a promising target, with tyrosine kinase inhibitors (TKIs) approved for the treatment of lung cancer. However, their efficacy is limited compared to other targeted therapies.
Our goal is to improve therapies targeting MET through three main areas:
- understanding the mechanisms of oncogenic addiction
- identifying resistance mechanisms
- expanding patient eligibility for targeted therapies.
To achieve this, our team focuses on lung and prostate cancers, the main causes of cancer mortality. Our research provides fundamental knowledge on oncogenic addiction and proposes new therapeutic strategies.
We have deliberately chosen to develop translational research and to work closely with molecular biologists, pathologists and clinicians at the CHU, who are directly involved in patient care.
Our published and ongoing projects include:
Control of cell death/survival balance by the MET dependence receptor. Duplaquet L, Leroy C, Vinchent A, Paget S, Lefebvre J, Vanden Abeele F, Lancel S, Giffard F, Bidaux G, Heliot L, Poulain L, Furlan A and Tulasne D. eLife. 2020 https://doi.org/10.7554/elife.50041
Non-small cell lung cancer (NSCLC) is the archetype of cancers treated by targeted therapies. Mutations of the MET receptor affecting the splicing sites of exon 14 lead to the deletion of a regulatory domain (METex14Del). Using knock-in mice, we demonstrated that this domain contains a caspase cleavage site involved in amplifying apoptosis. Thus, MET can act as both an oncogene and a tumor suppressor.
MET exon 14 skipping mutation is a hepatocyte growth factor (HGF)-dependent oncogenic driver in vitro and in humanised HGF knock-in mice. Fernandes M, Hoggard B, Jamme P, Paget S, Truong MJ, Grégoire V, Vinchent A, Descarpentries C, Morabito A, Stanislovas J, Farage E, Meneboo JP, Sebda S, Bouchekioua-Bouzaghou K, Nollet M, Humez S, Perera T, Fromme P, Grumolato L, Figeac M, Copin MC, Tulasne D, Cortot AB, Kermorgant S, Kherrouche Z. Mol Oncol. 2023 https://doi.org/10.1002/1878-0261.13397
By recreating the METex14Del mutation through genome editing and using humanized mice for HGF, the MET ligand, we showed that transformation by METex14Del still depends on its stimulation. This suggests that the ligand could serve as a biomarker to identify responsive patients.
Alterations in the PI3K pathway drive resistance to MET inhibitors in NSCLC harboring MET exon 14 skipping mutations. Jamme P, Fernandes M, Copin MC, Descarpentries C, Escande F, Morabito A, Grégoire V, Jamme M, Baldacci S, Tulasne D, Kherrouche Z* and Cortot AB*. J Thorac Oncol. 2020 https://doi.org/10.1016/j.jtho.2020.01.027
The Lille University Hospital contributed to international clinical trials demonstrating the efficacy of TKIs against METex14Del in NSCLC. However, resistance to TKIs has been observed. Using patient-derived cell lines, we identified one of the first resistance mechanisms, involving the PI3K pathway.
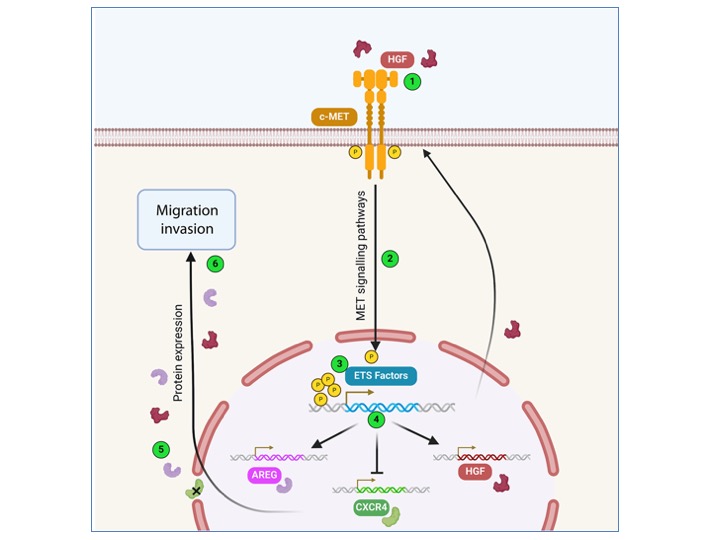
Functional interaction between receptor tyrosine kinase MET and ETS transcription factors promotes prostate cancer progression. Carouge E, Burnichon C, Figeac M, Sebda S, Vanpouille N, Vinchent A, Truong MJ, Duterque-Coquillaud M, Tulasne D and Chotteau-Lelièvre A. Mol Oncol. 2024, http://doi.org/10.1002/1878-0261.13739
Castration-resistant prostate cancer (CRPC) is not eligible for targeted therapies, but MET is re-expressed in these tumors. Moreover, nearly half of these cancers overexpress ETS transcription factors due to chromosomal translocations. We demonstrated that MET and ETS factors cooperate to induce tumor growth. This response is inhibited by MET-targeting TKIs, paving the way for targeted therapies in these cancers.
Fascin-1 expression is associated with neuroendocrine prostate cancer and directly suppressed by androgen receptor. Turpin A*, Delliaux C*, Parent P, Chevalier H, Escudero-Iriarte C, Bonardi F, Vanpouille N, Flourens A, Querol J, Carnot A, Leroy X, Herranz N, Lanel T, Villers A, Olivier J, Touzet H, de Launoit Y, Tian TV, Duterque-Coquillaud M. Br J Cancer. 2023 https://www.nature.com/articles/s41416-023-02449-x.pdf
We showed that Fascin-1, an actin bundling protein involved in invadopodia formation, is overexpressed during prostate cancer progression into neuroendocrine (NEPC), an aggressive form of CRPC. We identified Fascin-1 gene as an AR-repressed gene, therefore its repression is lost in these neuroendocrine cancers characterized by the absence or inactivity of AR. Since Fascin-1 is involved in NEPC progression and aggressiveness, our results provide the rationale for the future clinical development of Fascin-1 inhibitors in NEPC patients, which could represent a promising targeted therapy.
2024
Carouge E, Burnichon C, Figeac M, Sebda S, Vanpouille N, Vinchent A, Truong MJ, Duterque-Coquillaud M, Tulasne D, Chotteau-Leliévre A. Functional interaction between receptor tyrosine kinase MET and ETS transcription factors promotes prostate cancer progression. Molecular Oncology, 2024, PMID: 39374163, http://doi.org/10.1002/1878-0261.13739
Guérin C, Tulasne D. Recording and classifying MET receptor mutations in cancers. Elife. 2024 Apr 23;13:e92762. doi: 10.7554/eLife.92762. PMID: 38652103 Free PMC article. Review.
2023
Turpin A, Delliaux C, Parent P, Chevalier H, Escudero-Iriarte C, Bonardi F, Vanpouille N, Flourens A, Querol J, Carnot A, Leroy X, Herranz N, Lanel T, Villers A, Olivier J, Touzet H, de Launoit Y, Tian TV, Duterque-Coquillaud M. Fascin-1 expression is associated with neuroendocrine prostate cancer and directly suppressed by androgen receptor. Br J Cancer. 2023 Dec;129(12):1903-1914. doi: 10.1038/s41416-023-02449-x. Epub 2023 Oct 24. PMID: 37875732
Dehem A, Mazieres J, Chour A, Guisier F, Ferreira M, Boussageon M, Girard N, Moro-Sibilot D, Cadranel J, Zalcman G, Ricordel C, Wislez M, Munck C, Poulet C, Gauvain C, Descarpentries C, Wasielewski E, Cortot AB, Baldacci S. Characterization of 164 patients with NRAS mutated non-small cell lung cancer (NSCLC). Lung Cancer. 2023 Oct 9;186:107393. doi: 10.1016/j.lungcan.2023.107393. Online ahead of print.PMID: 37839252
Chour A, Denis J, Mascaux C, Zysman M, Bigay-Game L, Swalduz A, Gounant V, Cortot A, Darrason M, Fallet V, Auclin E, Basse C, Tissot C, Decroisette C, Bombaron P, Giroux-Leprieur E, Odier L, Brosseau S, Creusot Q, Gueçamburu M, Meersseman C, Rochand A, Costantini A, Gaillard CM, Wasielewski E, Girard N, Cadranel J, Lafitte C, Lebossé F, Duruisseaux M. Brief Report: Severe Sotorasib-Related Hepatotoxicity and Non-Liver Adverse Events Associated With Sequential Anti-Programmed Cell Death (Ligand)1 and Sotorasib Therapy in KRASG12C-Mutant Lung Cancer. J Thorac Oncol. 2023 Oct;18(10):1408-1415. doi: 10.1016/j.jtho.2023.05.013. Epub 2023 May 20. PMID: 37217096.
Khiter F, Kherrouche Z, Dubois V, Slupek S, Petit E, Debrie AS, Cauchi S, Barois N, Rouanet C, Mielcarek N. Combined regulation of pro-inflammatory cytokines production by STAT3 and STAT5 in a model of B. pertussis infection of alveolar macrophages. Front Immunol. 2023 Sep 28;14:1254276. doi: 10.3389/fimmu.2023.1254276. eCollection 2023.
Ardin C, Humez S, Leroy V, Ampere A, Bordier S, Escande F, Turlotte A, Stoven L, Nunes D, Cortot A, Gauvain C. Pursuit or discontinuation of anti-PD1 after 2 years of treatment in long-term responder patients with non-small cell lung cancer. Ther Adv Med Oncol. 2023 Sep 13;15:17588359231195600. doi: 10.1177/17588359231195600.
Mazieres J, Paik PK, Garassino MC, Le X, Sakai H, Veillon R, Smit EF, Cortot AB, Raskin J, Viteri S, Wu YL, Yang JCH, Ahn MJ, Ma R, Zhao J, O’Brate A, Berghoff K, Bruns R, Otto G, Johne A, Felip E, Thomas M. Tepotinib Treatment in Patients With MET Exon 14-Skipping Non-Small Cell Lung Cancer: Long-term Follow-up of the VISION Phase 2 Nonrandomized Clinical Trial. JAMA Oncol. 2023 Sep 1;9(9):1260-1266. doi: 10.1001/jamaoncol.2023.1962. PMID: 37270698.
Fernandes M, Paget S, Kherrouche Z, Truong MJ, Vinchent A, Meneboo JP, Sebda S, Werkmeister E, Descarpentries C, Figeac M, Cortot AB and Tulasne. D. Transforming properties of MET receptor exon 14 skipping can be recapitulated by loss of the CBL ubiquitin ligase binding site. FEBS Lett. 2023 Jul 19. doi: 10.1002/1873-3468.14702.
Delval L, Hantute-Ghesquier A, Sencio V, Flaman JM, Robil C, Angulo FS, Lipskaia L, Çobanoğlu O, Lacoste AS, Machelart A, Danneels A, Corbin M, Deruyter L, Heumel S, Idziorek T, Séron K, Sauve F, Bongiovanni A, Prévot V, Wolowczuk I, Belouzard S, Saliou JM, Gosset P, Bernard D, Rouillé Y, Adnot S, Duterque-Coquillaud M, Trottein F. Removal of senescent cells reduces the viral load and attenuates pulmonary and systemic inflammation in SARS-CoV-2-infected, aged hamsters. Nat Aging. 2023 Jul;3(7):829-845. doi: 10.1038/s43587-023-00442-w.
Tsuboi M, Herbst RS, John T, Kato T, Majem M, Grohé C, Wang J, Goldman JW, Lu S, Su WC, de Marinis F, Shepherd FA, Lee KH, Le NT, Dechaphunkul A, Kowalski D, Poole L, Bolanos A, Rukazenkov Y, Wu YL; ADAURA Investigators. Overall Survival with Osimertinib in Resected EGFR-Mutated NSCLC. N Engl J Med. 2023 Jul 13;389(2):137-147. doi: 10.1056/NEJMoa2304594. Epub 2023 Jun 4.PMID: 37272535.
L Brunet, D Alexandre, J Lee, MM Blanquer-Rossello, A Guernet, H. Chhouri, Z. Kherrouche, A. Arabo, S. Yao, D. Godefroy, J. Dehedin, J. Li, C. Duparc, P. Jamme, A. Vinchent, C. Berard, D. Tulasne, S. Arena, A. Bardelli, C. Cheng, B. Chul Cho, C. Coulouarn, S. Aaronson, AB. Cortot, Y. Anouar, L. Grumolato. Prolonging lung cancer response to EGFR inhibition by targeting the selective advantage of resistant cells. bioRxiv, 2023.06. 19.545595
Baudry AS, Charton E, Piessen G, Vanlemmens L, Cortot A, Ceban T, Anota A, Christophe V. Emotional distress, supportive care needs and age in the prediction of quality of life of cancer patients’ caregivers: A cross-sectional study. Eur J Oncol Nurs. 2023 Jun;64:102324. doi: 10.1016/j.ejon.2023.102324.
Fernandes M, Hoggard B, Jamme P, Paget S, Truong MJ, Grégoire V, Vinchent A, Descarpentries C, Morabito A, Stanislovas J, Farage E, Meneboo JP, Sebda S, Bouchekioua-Bouzaghou K, Nollet M, Humez S, Perera T, Fromme P, Grumolato L, Figeac M, Copin MC, Tulasne D, Cortot AB, Kermorgant S, Kherrouche Z. MET exon 14 skipping mutation is a hepatocyte growth factor (HGF)-dependent oncogenic driver in vitro and in humanised HGF knock-in mice. Mol Oncol. 2023 Feb 17. doi: 10.1002/1878-0261.13397.
Bogard G, Barthelemy J, Hantute-Ghesquier A, Sencio V, Brito-Rodrigues P, Séron K, Robil C, Flourens A, Pinet F, Eberlé D, Trottein F, Duterque-Coquillaud M, Wolowczuk I. SARS-CoV-2 infection induces persistent adipose tissue damage in aged golden Syrian hamsters. Cell Death Dis. 2023 Feb 1;14(2):75. doi: 10.1038/s41419-023-05574-w.
2022
Mushtaq R, Cortot AB, Gautschi O, Mazieres J, Camidge DR. PD-1/PD-L1 inhibitor activity in patients with gene-rearrangement positive non-small cell lung cancer-an IMMUNOTARGET case series. Transl Lung Cancer Res. 2022 Dec;11(12):2412-2417. doi: 10.21037/tlcr-22-329.
Christophe V, Anota A, Vanlemmens L, Cortot AB, Ceban T, Piessen G, Charton E, Baudry AS. Unmet supportive care needs of caregivers according to medical settings of cancer patients: a cross-sectional study. Support Care Cancer. 2022 Nov;30(11):9411-9419. doi: 10.1007/s00520-022-07379-7.
Debieuvre D, Molinier O, Falchero L, Locher C, Templement-Grangerat D, Meyer N, Morel H, Duval Y, Asselain B, Letierce A, Trédaniel J, Auliac JB, Bylicki O, Moreau L, Fore M, Corre R, Couraud S, Cortot AB; Study Group KBP-2020-CPHG; KBP-2020-CPHG. Lung cancer trends and tumor characteristic changes over 20 years (2000-2020): Results of three French consecutive nationwide prospective cohorts’ studies. Lancet Reg Health Eur. 2022 Aug 29;22:100492. doi: 10.1016/j.lanepe.2022.100492.
Veillon R, Sakai H, Le X, Felip E, Cortot AB, Smit EF, Park K, Griesinger F, Britschgi C, Wu YL, Melosky B, Baijal S, Jr GC, Sedova M, Berghoff K, Otto G, Paik PK. Safety of Tepotinib in Patients With MET Exon 14 Skipping NSCLC and Recommendations for Management. Clin Lung Cancer. 2022 Jun;23(4):320-332. doi: 10.1016/j.cllc.2022.03.002.
Baldacci S, Besse B, Avrillon V, Mennecier B, Mazieres J, Dubray-Longeras P, Cortot AB, Descourt R, Doubre H, Quantin X, Duruisseaux M, Monnet I, Moro-Sibilot D, Cadranel J, Clément-Duchêne C, Cousin S, Ricordel C, Merle P, Otto J, Schneider S, Langlais A, Morin F, Westeel V, Girard N. Lorlatinib for advanced anaplastic lymphoma kinase-positive non-small cell lung cancer: Results of the IFCT-1803 LORLATU cohort. Eur J Cancer. 2022 May;166:51-59. doi: 10.1016/j.ejca.2022.01.018.
Cortot AB, Le X, Smit E, Viteri S, Kato T, Sakai H, Park K, Camidge DR, Berghoff K, Vlassak S, Paik PK. Safety of MET Tyrosine Kinase Inhibitors in Patients With MET Exon 14 Skipping Non-small Cell Lung Cancer: A Clinical Review. Clin Lung Cancer. 2022 May;23(3):195-207. doi: 10.1016/j.cllc.2022.01.003.
Sebai M, Tulasne D, Caputo SM, Verkarre V, Fernandes M, Guérin C, Reinhart F, Adams S, Maugard C, Caron O, Guillaud-Bataille M, Berthet P, Bignon YJ, Bressac-de Paillerets B, Burnichon N, Chiesa J, Giraud S, Lejeune S, Limacher JM, de Pauw A, Stoppa-Lyonnet D, Zattara-Cannoni H, Deveaux S, Lidereau R, Richard S, Rouleau E. Novel germline MET pathogenic variants in French patients with papillary renal cell carcinomas type I. Hum Mutat. 2022 Mar;43(3):316-327. doi: 10.1002/humu.24313.
Sencio V, Machelart A, Robil C, Benech N, Hoffmann E, Galbert C, Deryuter L, Heumel S, Hantute-Ghesquier A, Flourens A, Brodin P, Infanti F, Richard V, Dubuisson J, Grangette C, Sulpice T, Wolowczuk I, Pinet F, Prévot V, Belouzard S, Briand F, Duterque-Coquillaud M, Sokol H, Trottein F. Alteration of the gut microbiota following SARS-CoV-2 infection correlates with disease severity in hamsters. Gut Microbes. 2022 Jan-Dec;14(1):2018900. doi: 10.1080/19490976.2021.2018900.
2021
Palma M, Leroy C, Salomé-Desnoulez S, Werkmeister E, Kong R, Mongy M, Le Hir H, Lejeune F. A role for AKT1 in nonsense-mediated mRNA decay. Nucleic Acids Res. 2021 Nov 8;49(19):11022-11037. doi: 10.1093/nar/gkab882.
Le X, Sakai H, Felip E, Veillon R, Garassino MC, Raskin J, Cortot AB, Viteri S, Mazieres J, Smit EF, Thomas M, Iams WT, Cho BC, Kim HR, Yang JC, Chen YM, Patel JD, Bestvina CM, Park K, Griesinger F, Johnson M, Gottfried M, Britschgi C, Heymach J, Sikoglu E, Berghoff K, Schumacher KM, Bruns R, Otto G, Paik PK. Tepotinib Efficacy and Safety in Patients with MET Exon 14 Skipping NSCLC: Outcomes in Patient Subgroups from the VISION Study with Relevance for Clinical Practice. Clin Cancer Res. 2021 Nov 17. doi: 10.1158/1078-0432.CCR-21-2733.
Vary A, Lebellec L, Di Fiore F, Penel N, Cheymol C, Rad E, El Hajbi F, Lièvre A, Edeline J, Bimbai AM, Le Deley MC, Turpin A. FOLFIRINOX relative dose intensity and disease control in advanced pancreatic adenocarcinoma. Ther Adv Med Oncol. 2021 Jul 16;13:17588359211029825. doi: 10.1177/17588359211029825.
Pinquie F, Cortot AB, Chevalier LM, Morel A, Sandrini J, Guguen C, Morvan B, Molinier O. A Case Report of Successful Treatment With Crizotinib to Overcome Resistance to Osimertinib in an EGFR Mutated Non-Small-Cell Lung Cancer Patient Harboring an Acquired MET Exon 14 Mutation. Clin Lung Cancer. 2021 Jun 13:S1525-7304(21)00142-X. doi: 10.1016/j.cllc.2021.06.002.
Cortot AB, Madroszyk A, Giroux-Leprieur E, Molinier O, Quoix E, Bérard H, Otto J, Rault I, Moro-Sibilot D, Raimbourg J, Amour E, Morin F, Hureaux J, Moreau L, Debieuvre D, Morel H, Renault A, Pichon E, Huret B, Charpentier S, Denis MG, Cadranel J. First-Line Afatinib plus Cetuximab for EGFR-Mutant Non-Small Cell Lung Cancer: Results from the Randomized Phase II IFCT-1503 ACE-Lung Study. Clin Cancer Res. 2021 Aug 1;27(15):4168-4176. doi: 10.1158/1078-0432.CCR-20-4604.
Fernandes M, Jamme P, Cortot AB, Kherrouche Z, Tulasne D. When the MET receptor kicks in to resist targeted therapies. Oncogene. 2021 Jun;40(24):4061-4078. doi: 10.1038/s41388-021-01835-0.
Jeschke J, Collignon E, Al Wardi C, Krayem M, Bizet M, Jia Y, Garaud S, Wimana Z, Calonne E, Hassabi B, Morandini R, Deplus R, Putmans P, Dube G, Singh NK, Koch A, Shostak K, Rizzotto L, Ross RL, Desmedt C, Bareche Y, Rothé F, Lehmann-Che J, Duterque-Coquillaud M, Leroy X, Menschaert G, Teixeira L, Guo M, Limbach PA, Close P, Chariot A, Leucci E, Ghanem G, Yuan BF, Willard-Gallo K, Sotiriou C, Marine JC, Fuks F. Downregulation of the FTO m6A RNA demethylase promotes EMT-mediated progression of epithelial tumors and sensitivity to Wnt inhibitors. Nat Cancer. 2021 Jun;2(6):611-628. doi: 10.1038/s43018-021-00223-7.
Vauléon E, Behal H, Lebellec L, Desbarbieux R, Baldacci S, Simon N, Pannier D, Vieillard MH, Turpin A. Does bevacizumab increase joint pain in patients with cancer? Results of the prospective observational BEVARTHRALGIA study. Cancer Chemother Pharmacol. 2021 Apr;87(4):533-541. doi: 10.1007/s00280-020-04226-6.
Palma M, Lejeune F. Deciphering the molecular mechanism of stop codon readthrough. Biol Rev Camb Philos Soc. 2021 Feb;96(1):310-329. doi: 10.1111/brv.12657. Epub 2020 Oct 22. PMID: 33089614.
2020
Paik PK, Felip E, Veillon R, Sakai H, Cortot AB, Garassino MC, Mazieres J, Viteri S, Senellart H, Van Meerbeeck J, Raskin J, Reinmuth N, Conte P, Kowalski D, Cho BC, Patel JD, Horn L, Griesinger F, Han JY, Kim YC, Chang GC, Tsai CL, Yang JC, Chen YM, Smit EF, van der Wekken AJ, Kato T, Juraeva D, Stroh C, Bruns R, Straub J, Johne A, Scheele J, Heymach JV, Le X. Tepotinib in Non-Small-Cell Lung Cancer with MET Exon 14 Skipping Mutations. N Engl J Med. 2020 May 29. doi: 10.1056/NEJMoa2004407.
Bouchet M, Lainé A, Boyault C, Proponnet-guerault M, Meugnier E, Bouazza l, Kan C, Geraci S, El moghrabi S, Hernandez-vargas MH, Benetollo C, Yoshiko Y, Duterque-coquillaud M, Clézardin P, Marie JC and Bonnelye E. ERRα Expression in Bone Metastases Leads to an Exacerbated Antitumor Immune Response. Cancer research, 2020 Jul 1;80(13):2914-2926. doi: 10.1158/0008-5472.CAN-19-3584.
Thureau S, Faivre JC, Assaker R, Biver E, Confavreux CB, Debiais F, Duterque-coquillaud M, Giammaril F, Heymann D, Lecouvet FE, Morardet I, Paycha F, Body JJ, Vieillard MH. Adapting palliative radiation therapy for bone metastases during the Covid-19 pandemic: GEMO position paper. Journal of bone oncology, 2020 Jun; 22: 100291. doi: 10.1016/j.jbo.2020.100291.
Cortot AB, Audigier-Valette C, Molinier O, Le Moulec S, Barlesi F, Zalcman G, Dumont P, Pouessel D, Poulet C, Fontaine-Delaruelle C, Hiret S, Dixmier A, Renault PA, Becht C, Raffy O, Dayen C, Mazieres J, Pichon E, Langlais A, Morin F, Moro-Sibilot D, Besse B. Weekly paclitaxel plus bevacizumab versus docetaxel as second- or third-line treatment in advanced non-squamous non-small-cell lung cancer: Results of the IFCT-1103 ULTIMATE study. Eur J Cancer. 2020 May;131:27-36.
Marquette CH, Boutros J, Benzaquen J, Ferreira M, Pastre J, Pison C, Padovani B, Bettayeb F, Fallet V, Guibert N, Basille D, Ilie M, Hofman V, Hofman P; AIR project Study Group. Circulating tumour cells as a potential biomarker for lung cancer screening: a prospective cohort study. Lancet Respir Med. 2020 Jul;8(7):709-716. doi: 10.1016/S2213-2600(20)30081-3. PMID: 32649919.
Domblides C, Leroy K, Monnet I, Mazières J, Barlesi F, Gounant V, Baldacci S, Mennecier B, Toffart AC, Audigier-Valette C, Doucet L, Giroux-Leprieur E, Guisier F, Ricordel C, Molinier O, Perol M, Pichon E, Robinet G, Templement-Grangerat D, Ruppert AM, Rabbe N, Antoine M, Wislez M. Efficacy of Immune Checkpoint Inhibitors in Lung Sarcomatoid Carcinoma. J Thorac Oncol. 2020 May;15(5):860-866. doi: 10.1016/j.jtho.2020.01.014. Epub 2020 Jan 25. PMID: 31991225.
Jamme P, Fernandes M, Copin MC, Descarpentries C, Escande F, Morabito A, Grégoire V, Jamme M, Baldacci S, Tulasne D, Kherrouche Z* and Cortot AB*. Alterations in the PI3K pathway drive resistance to MET inhibitors in NSCLC harboring MET exon 14 skipping mutations. Journal of Thoracic Oncology. 2020 May;15(5):741.
Trzaska C, Amand S, Bailly C, Leroy C, Marchand V, Duvernois-Berthet E, Saliou JM, Benhabiles H, Werkmeister E, Chassat T, Guilbert R, Hannebique D, Mouray A, Copin MC, Moreau PA, Adriaenssens E, Kulozik A, Westhof E, Tulasne D, Motorin Y, Rebuffat S and Lejeune F. 2,6-Diaminopurine as a highly potent corrector of UGA nonsense mutations. Nat Commun. 2020 Mar 20;11(1):1509. doi: 10.1038/s41467-020-15140-z.
Turpin A, Duterque−coquillaud M, Vieillard MH. Bone metastasis: current state of play. Translational oncology. 2020 feb;13(2):308-320. IF 3.138
Baldacci S, Figeac M, Antoine M, Descarpentries C, Kherrouche Z, Jamme P, Copin MC, Tulasne D, Nanni I, Beau-Faller M, Melaabi S, Levallet G, Quoix E, Moro-Sibilot D, Friard S, Missy P, Barlesi F, Cadranel J, and Cortot AB. High MET Overexpression Does Not Predict the presence of MET exon 14 Splice Mutations in NSCLC: Results From the IFCT PREDICT.amm study. Journal of Thoracic Oncology. 2020 Jan;15(1):120-124.
Bennouna J, Girard N, Audigier-Valette C, le Thuaut A, Gervais R, Masson P, Marcq M, Molinier O, Cortot A, Debieuvre D, Cadranel J, Lena H, Moro-Sibilot D, Chouaid C, Mennecier B, Urban T, Sagan C, Perrier L, Barlesi F, Denis MG.Phase II Study Evaluating the Mechanisms of Resistance on Tumor Tissue and Liquid Biopsy in Patients With EGFR-mutated Non-pretreated Advanced Lung Cancer Receiving Osimertinib Until and Beyond Radiologic Progression: The MELROSE Trial. Clin Lung Cancer. 2020 Jan;21(1):e10-e14.
Besse B, Garrido P, Cortot AB, Johnson M, Murakami H, Gazzah A, Gil M, Bennouna J. Efficacy and safety of necitumumab and pembrolizumab combination therapy in patients with Stage IV non-small cell lung cancer. Lung Cancer. 2020 Apr;142:63-69.
Mazieres J, Barlesi F, Rouquette I, Molinier O, Besse B, Monnet I, Audigier-Valette C, Toffart AC, Renault PA, Fraboulet S, Hiret S, Mennecier B, Debieuvre D, Westeel V, Masson P, Madroszyk-Flandin A, Pichon E, Cortot AB, Amour E, Morin F, Zalcman G, Moro-Sibilot D, Souquet PJ. Randomized Phase II Trial Evaluating Treatment with EGFR-TKI Associated with Antiestrogen in Women with Nonsquamous Advanced-Stage NSCLC: IFCT-1003 LADIE Trial. Clin Cancer Res. 2020 Mar 6.
Duplaquet L, Leroy C, Vinchent A, Paget S, Lefebvre J, Vanden Abeele F, Lancel S, Giffard F, Bidaux G, Heliot L, Poulain L, Furlan A and Tulasne D. Control of cell death/survival balance by the MET dependence receptor. eLife. 2020 Feb 24;9:e50041. doi: 10.7554/eLife.50041.
2019
Duplaquet L, Figeac M, Leprêtre F, Frandemiche C, Villenet C, Sebda S, Sarafan-Vasseur N, Bénozène M, Vinchent A, Goormachtigh G, Wicquart W, Rousseau R, Beaussire L, Truant S, Michel P, Sabourin JC, Galateau F, Copin MC, Zalcman G, De Launoit Y, Fafeur V and Tulasne D. Functional analysis of somatic mutations affecting receptor tyrosine kinase family in metastatic colorectal cancer. Mol Cancer Ther. 2019 Mar 29. pii: molcanther.0582.2018.
Jamme P, Descarpentries C, Gervais R, Dansin E, Wislez M, Grégoire V, Richard N, Baldacci S, Rabbe N, Kyheng M, Kherrouche Z, Escande F, Copin MC, Cortot AB. Relevance of Detection of Mechanisms of Resistance to ALK Inhibitors in ALK-Rearranged NSCLC in Routine Practice. Clin Lung Cancer. 2019 Jul;20(4):297-304.
Turpin A, Duterque-Coquillaud M, Vieillard MH. Bone Metastasis: Current State of Play. Transl Oncol. 2019 Dec 23;13(2):308-320. Review. IF 3.138
Fernandes M, Duplaquet L, Tulasne D. Proteolytic cleavages of MET: the divide-and-conquer strategy of a receptor tyrosine kinase. BMB Rep. 2019 Apr;52(4):239-249. Review.
El Amrani M, Corfiotti F, Corvaisier M, Vasseur R, Fulbert M, Skrzypczyk C, Deshorgues AC, Gnemmi V, Tulasne D, Lahdaoui F, Vincent A, Pruvot FR, Van Seuningen I, Huet G, Truant S. Gemcitabine-induced epithelial-mesenchymal transition-like changes sustain chemoresistance of pancreatic cancer cells of mesenchymal-like phenotype. Mol Carcinog. 2019 Aug 2.
Gnangnon B, Fréville A, Cailliau K, Leroy C, De Witte C, Tulasne D, Martoriarti A, Jung V, Guerrera IC, Marion S, Khalife J, Pierrot C. Plasmodium pseudo-Tyrosine Kinase-like binds PP1 and SERA5 and is exported to host erythrocytes. Sci Rep. 2019 May 31;9(1):8120.
Vargas G, Bouchet M, Bouazza L, Reboul P, Boyault C, Gervais M, Kan C, Benetollo C, Brevet M, Croset M, Mazel M, Cayrefourcq L, Geraci S, Vacher S, Pantano F, Filipits M, Driouch K, Bieche I, Gnant M, Jacot W, Aubin JE, Duterque-Coquillaud M, Alix-Panabières C, Clézardin P, Bonnelye E. ERRα promotes breast cancer cell dissemination to bone by increasing RANK expression in primary breast tumors. Oncogene. 2019 Feb;38(7):950-964.
Pasquier D, Le Deley M, Tresch E, Cormier L, Duterque M, Nenan S, Lartigau E. GETUG-AFU 31: a phase I/II multicentre study evaluating the safety and efficacy of salvage stereotactic radiation in patients with intraprostatic tumour recurrence after external radiation therapy-study protocol. BMJ Open. 2019 Aug 2;9(8):e026666.
2018
[Delliaux C, Tian TV], Bouchet M, Fradet A, Vanpouille N, Flourens A, Deplus R, Villers A, Leroy X, Clézardin P, de Launoit Y, Bonnelye E, Duterque-Coquillaud M. TMPRSS2:ERG gene fusion expression regulates bone markers and enhances the osteoblastic phenotype of prostate cancer bone metastases. Cancer Lett. 2018 Dec 1;438:32-43.
Dumortier M, Ladam F, Damour I, Vacher S, Bieche I, Marchand N, DeLaunoit Y, Tulasne D and Chotteau-Lelievre A. ETV4 transcription factor and MMP13 metalloprotease are interplaying actors of breast tumorigenesis. Breast Cancer Res. 2018 Jul 11;20(1):73.
Descarpentries C, Leprêtre F, Escande F, Kherrouche Z, Figeac M, Sebda S, Baldacci S, Grégoire V, Jamme P, Copin MC, Tulasne D and Cortot AB. Optimization of routine testing for MET exon 14 splice site mutations in non-small cell lung cancer patients. Journal of Thoracic Oncology, 2018 Sep 6. pii: S1556-0864(18)33038-7.
[Baldacci S, Kherrouche Z], Cockenpot V, Stoven L, , Copin M.C, Werkmeister E, Marchand N, Maéva Kyheng, Tulasne D and. Cortot A.B. MET amplification increases the metastatic spread of EGFR-mutated NSCLC. Lung Cancer, 2018 (125)57-67.
Mekki S.M, Mougel A, Vinchent A, Paquet C, Copin M.C, Leroy L, Kherrouche Z, Bonte J.P, Melnyk O, Vicogne J. and Tulasne D. Hypoxia leads to decreased autophosphorylation of the MET receptor and promotes its resistance to tyrosine kinase inhibitors. Oncotarget. 2018 Jun 5;9(43):27039-27058.
Duplaquet L, Kherrouche Z, Baldacci S, Jamme P, Cortot AB, Copin MC and Tulasne D. The multiple paths towards MET receptor addiction in cancer. Oncogene. 2018 Jun; 37(24):3200-3215. Review.
Baldacci S, Kherrouche Z, Descarpentries C, Wislez M, Dansin E, Furlan A, Tulasne D, Cortot AB. MET exon 14 splicing sites mutations: A new therapeutic opportunity in lung cancer. Rev Mal Respir. 2018 Aug 31. pii: S0761-8425(18)30200-6. Review.
Hasne J, Hague F, Rodat-despoix L, Geerts D, Leroy C, Tulasne D, Ouadid-Ahidouh H and Kischpel P. Orai3 calcium channel and resistance to chemotherapy in breast cancer cells: the p53 connection. Cell Death Differ. 2018 Jan 11.
A Bokhari, V Jonchere, A Lagrange, R Bertrand, M Svrcek, L Marisa, O Buhard, M Greene, A Demidova, J Jia, E Adriaenssens, T Chassat, DS Biard, JF Flejou, F Lejeune, A Duval, A Collura. Targeting nonsense-mediated mRNA decay in colorectal cancers with microsatellite instability. Oncogenesis. 2018 Sep 19; 7(9):70;
2017
Deplus R, Delliaux C, Marchand N, Flourens A, Vanpouille N, Leroy X, de Launoit Y, Duterque-Coquillaud M. TMPRSS2-ERG fusion promotes prostate cancer metastases in bone. Oncotarget. 2017 Feb 14;8(7):11827-11840.
Jia J, Gonzalez-Hilarion S, Werkmeister E, Lafont F, Grunert D.C., Tulasne D and. Lejeune F. PTC readthrough in human cells occurs in novel cytoplasmic foci and requires UPF proteins. J Cell Sci. 2017 Jul 25. pii: jcs.198176.
Benhabiles H, Gonzalez-Hilarion S, Amand S, Bailly C, Prévotat A, Reix P, Hubert D, Adriaenssens E, Rebuffat S, Tulasne D, Lejeune F. Optimized approach for the identification of highly efficient correctors of nonsense mutations in human diseases. PLoS One. 2017 Nov 13;12(11):e0187930.
Baldacci S, Mazieres J, Tomasini P, Girard N, Guisier F, Audigier-Valette C, Monnet I, Wislez M, Pérol M, Dô P, Dansin E, Leduc C, Leprieur EG, Moro-Sibilot D, Tulasne D, Kherrouche Z, Labreuche J, Cortot AB. Outcome of EGFR-mutated NSCLC patients with MET-driven resistance to EGFR tyrosine kinase inhibitors. Oncotarget. 2017 Oct 9;8(62):105103-105114.
Lapère C, Cortot A.B, Grégoire V, Cockenpot V, Tulasne D, and Copin M.C. Preferential localization of MET expression at the invasion front and in spreading cells through alveolar spaces in non-small-cell lung carcinomas. Am J Surg Pathol. 2017 Mar;41(3):414-422.
Montagne R, Baranzelli A, Muharram G, Leroy C, Debreuck N, Kherrouche Z, Lemière A, Cortot AB and Tulasne D. Calpain cleavage of the Met receptor variant R970C generates fragment promoting epithelial cells scattering. Oncotarget. 2017 Jan 4.
Cortot A.B, Kherrouche Z, Descarpentries C, Wislez M, Baldacci S, Furlan A and Tulasne D. Exon 14 deleted MET receptor as a new biomarker and target in cancers. J Natl Cancer Inst. 2017 May 1;109(5). Review.
Lejeune F. Nonsense-mediated mRNA decay at the crossroads of many cellular pathways. BMB Rep. 2017 Apr;50(4):175-185. Review
On-going theses
- TELLIER Rémi, D3 directed by David TULASNE.
- AMOUYEL Thomas, D3 co-directed by Martine DUTERQUE et Marie-Hélène VIEILLARD
- SEGUIER Denis, D2 directed by Jonathan OLIVIER
- ROUIS Maram, D1 directed by de David TULASNE
- BOUVIGNIES Léa, D1 co-directed by Martine DUTERQUE et Jonathan OLIVIER
- MARCQ Gautier, D4 directed by Arnaud VILLERS
Defended theses
- CAROUGE Elisa (February 2024) directed by Anne CHOTTEAU-LELIEVRE.
- GUERIN Célia (Decembrer2023) directed by David TULASNE.
- OLIVIER Jonathan (June 2021) directed by Arnaud VILLERS.
- PALMA Martine (October 2021) directed by Fabrice LEJEUNE
- FERNANDES Marie (April 2021) directed by David TULASNE.
- TURPIN Anthony (March 2021) directed by Martine DUTERQUE-COQUILLAUD.
- JAMME Philippe (January 2020) co-directed by Alexis CORTOT et Zoulika KHERROUCHE
- DUPLAQUET Leslie (2018) directed by David TULASNE
HDR
- OLIVIER Jonathan, 2024
