

Notre équipe « Sénescence, fibrose et cancer » (acronyme: SENFIB) est composée de chercheurs, professeurs associés, cliniciens joignant leur expertise, et leurs ressources pour développer des programmes de recherche fondamentaux et translationnels concernant l’impact des deux principales formes de vieillissement cellulaire (sénescence et fibrose) sur l’initiation du cancer ainsi que sur la récidive du cancer après la dormance.
Dans ce contexte, les objectifs de l’équipe (schématisés dans la figure suivante) seront de décrypter les interrelations entre sénescence et fibrose, d’analyser leur fonction sur la tumorigenèse dans le contexte du vieillissement et en réponse aux thérapies anti-cancéreuses et de rechercher de nouveaux séno-fibrolytiques qui pourraient éliminer ces cellules pour diminuer leurs impacts.
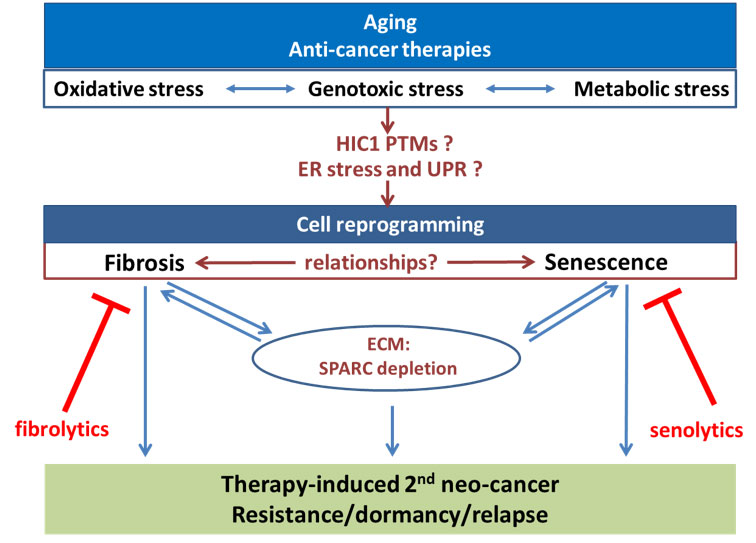
Notre recherche se concentre sur :
- La recherche de voies communes contrôlant la mise en place de la sénescence et de la fibrose
- Décrypter certains des mécanismes par lesquels la sénescence et la fibrose pourraient favoriser les premières étapes de la cancérogenèse
- Trouver de nouvelles cibles pour les séno-fibrolytiques parmi les ARN non codantsDans l’ensemble, notre équipe est impliquée dans de nouvelles connaissances fondamentales sur la régulation moléculaire de la sénescence et de la fibrose, deux états cellulaires associés à l’âge et induits par la thérapie anticancéreuse. Des projets interdisciplinaires avec des bioinformaticiens et des statisticiens permettront de développer de nouveaux modèles originaux des réseaux moléculaires. Notre projet fournira également des preuves de l’impact de ces états cellulaires reprogrammés sur l’initiation du cancer et la résistance des cellules cancéreuses à la thérapie par le biais de mécanismes autonomes de dormance des cellules tumorales ou par l’altération du microenvironnement. Des programmes translationels impliquant les cliniciens de l’équipe ainsi que des collaborations avec le Centre Oscar Lambret et OncoVet permettront d’évaluer le potentiel d’élimination des cellules sénescentes et/ou fibrotiques en tant que nouvelles thérapies pour empêcher l’initiation de deuxièmes néo-cancers post-traitement ou pour diminuer la résistance des cellules cancéreuses au traitement, l’entrée en dormance et la récurrence de la maladie.
Chaque année, nous accueillons 6 à 10 étudiants (L3, M1, Master 2, PhD) et post-doctorants au laboratoire (Technologie, Sciences de la Santé, Médecine et Pharmacie). Nous appartenons à l’école doctorale biologie-santé (http://edbsl.univ-lille.fr).
> CHERCHEURS
> INGÉNIEURS / TECHNICIENS
> ÉTUDIANTS
CHERCHEURS
 |
Pr Corinne ABBADIE, Professeur des Universités PU, PhD corinne.abbadie(@)ibl.cnrs.fr |
 |
Dr Yvan DE LAUNOIT, Directeur de Recherche CNRS, PhD Responsabilités collectives : Directeur Adjoint Scientifique INSB CNRS, Directeur adjoint ITMO Cancer, Porteur CPER Cancer. yvan.delaunoit(@)ibl.cnrs.fr |
 |
Pr François GLOWACKI, Nephrologue, MD PhD francois.glowacki(@)chru-lille.fr |
|
Pr Nicolas POTTIER, biologiste, PharmD PhD nicolas.pottier(@)univ-lille.fr |
 |
Pr Christelle CAUFFIEZ, Professeur des Universités PU, PhD christelle.cauffiez(@)univ-lille.fr |
 |
Pr Vanessa DEHENNAUT, Professeur des Universités PU, PhD Mot clés : colorectal cancer, therapy induced senescence, senescence escape, HBP, O-GlcNAcylation. vanessa.dehennaut(@)ibl.cnrs.fr
|
 |
Dr Albin POURTIER, Maître de conférences MCU, PhD albin.pourtier(@)ibl.cnrs.fr |
 |
Dr Olivier PLUQUET, Maître de conférences MCU, PhD olivier.pluquet(@)ibl.cnrs.fr |
INGÉNIEURS / TECHNICIENS
 |
Nathalie MARTIN : Ingénieur d’étude, CNRS nathalie.martin(@)cnrs.fr |
|
Ingrid LOISON : Assistant ingénieur CNRS ingrid.loison(@)ibl.cnrs.fr |
|
Dr Cynthia VAN DER HAUWAERT, PhD : Ingénieur de Recherche CHU cynthia.vanderhauwaert(@)inserm.fr |
|
Clémentine DE SCHUTTER : Technicienne de recherche, IPL clementine.de-schutter(@)ibl.cnrs.fr |
|
Nihad BOUKROUT : Ingénieur de Recherche Inserm nihad.boukrout@inserm.fr |
ÉTUDIANTS
|
Joëlle GIROUD (D4, doctorante, Lille/Namur) joelle.giroud(@)unamur.be |
|
Romain LARRUE (D4, doctorant) romain.larrue(@)chru-lille.fr |
|
Henry ABI RACHEB, Dermatologue, MD (D3, doctorant) henry.abiracheb(@)chu-lille.fr |
| |
Elodie RODZINSKI (D3, doctorante) elodie.rodzinski(@)ibl.cnrs.fr |
|
Emilie Combémorel (M2, Univ Lille) emilie.combemorel.etu(@)univ-lille.fr |
|
Adrien PIOGER (D1, doctorant) adrien.pioger.etu(@)univ-lille.fr |
|
Quentin BOURGERY (D1, doctorant) quentin.bourgery.etu(@)univ-lille.fr |
A- Mécanismes moléculaires de la sénescence cellulaire qui ont un impact sur la transformation néoplasique.
A.1- Sénescence et Émergence néoplasique post-sénescence
Le paradigme dominant dans le domaine de la sénescence est que la sénescence est un mécanisme suppresseur de tumeur que la cellule doit contourner pour devenir immortelle et tumorigène. À ce titre, il s’agit de la première ligne de défense induite suite à une activation oncogénique initiale. Les expériences que nous avons réalisées à l’aide de NHDFs ont confirmé ce concept. En effet, en culture in vitro, les NHDFs subissent une phase de croissance exponentielle suivie d’un plateau de sénescence, très stable, associé à des télomères raccourcis qui induisent une réponse permanente aux dommages à l’ADN (DDR) et un arrêt irréversible du cycle cellulaire (Nassour et al, Nature Commun, 2016). En revanche, les NHEKs subissent également une phase de croissance exponentielle suivie d’un plateau de sénescence, mais ce dernier n’est que transitoire et conduit à deux issues différents. La principale issue est la mort cellulaire qui affecte presque toutes les cellules. L’issue alternative affecte seulement environ une cellule sur 10 000. Ces cellules rentrent dans le cycle cellulaire et génèrent des clones de cellules qui prolifèrent à nouveau. Nous avons établi par des analyses transcriptomiques que ces cellules post-sénescence (PS) sont transformées (Martin et al, Cancer moléculaire, 2014) (Gosselin et al, Cancer Res, 2009). Nous avons donc appelé ce phénomène Emergence néoplasique post-sénescence (PSNE). Les NHEKs sénescentes et Post-Sénescentes (PS) affichent toujours de longs télomères, bien que la télomérase ne soit pas réactivée. Les cellules PS ne sont pas la descendance de cellules déjà transformées, ou cellules souches, qui auraient pu être présentes dans l’explant tissulaire d’origine, mais proviennent de la division de cellules mères pleinement sénescentes par un mécanisme atypique de mitose appelé néose (Gosselin et al, Cancer Res, 2009). La PSNE n’est pas spécifique des kératinocytes: nous avons décrit un phénomène similaire dans les cellules épithéliales mammaires normales (HMEC) (Nassour et al, Nature Commun, 2016).
Contact :
Corinne Abbadie, Corinne.abbadie@ibl.cnrs.fr
Olivier Pluquet, olivier.pluquet@ibl.cnrs.fr
A.2- L’émergence néoplasique post-sénescence procède en échappant à la mort cellulaire autophagique
En observant les cultures NHEK, nous avons émis l’hypothèse que les cellules sénescentes qui ne produisent pas de cellules PS meurent, ce qui suggère que la PSNE pourrait nécessiter un échappement à la mort cellulaire. Pour tester cette hypothèse, nous avons d’abord dû établir le mécanisme de la mort des cellules sénescentes qui était complètement inconnu. Nous avons démontré que les NHEKs sénescentes ne meurent pas par apoptose mais suite à une suractivation de l’autophagie (Gosselin et al, Am J Pathol. 2009). Nous avons poursuivi ce travail en recherchant ce qui pourrait induire cette activité autophagique élevée. Nous avons montré que la voie NF-kB> MnSOD> H2O2 dont nous avions démontré auparavant l’implication dans la survenue de sénescence dans les NHEKs (Bernard et al. Cancer Res 2004) était également responsable de l’induction de l’activité autophagique associée, via l’induction des dommages oxydatifs aux mitochondries et au noyau (Deruy et al. Plos One, 2010). Nous avons en outre démontré que l’inhibition de l’autophagie dans les NHEKs sénescents favorise la PSNE, démontrant formellement que la PSNE nécessite en effet un échappement à la mort cellulaire autophagique. Fait intéressant, nous avons également observé que les inhibiteurs de l’autophagie ont un effet de dose: à faibles doses, ils favorisent la PSNE, tandis qu’à fortes doses, ils ont tendance à la réprimer, ce qui suggère que les cellules sénescentes doivent maintenir une activité autophagique de contrôle de qualité pour pouvoir rentrer dans le cycle cellulaire (Deruy et al, Cell Death and Disease, 2014).
Contact :
Corinne Abbadie, Corinne.abbadie@ibl.cnrs.fr
A.3- L’émergence néoplasique post-sénescence se produit à la suite de cassures d’ADN simple brin non réparées
La réponse aux dommages à l’ADN (DDR) et l’activation de la voie p53 / p21 sont reconnues comme le principal mécanisme responsable de la robustesse et de l’irréversibilité de l’arrêt du cycle cellulaire à la sénescence. Elle est induite suite au raccourcissement des télomères ou suite à une activation oncogénique [2]. Dans le cas de la sénescence des NHEKs et HMECs, nous avons montré que cette voie n’est pas activée.
Au lieu de cela, la sénescence dans ces cellules épithéliales résulte d’un stress oxydatif léger, pas assez élevé pour induire des ruptures d’ADN double brin (DSB) et l’activation de la voie DDR, mais assez pour induire des ruptures d’ADN simple brin (SSB). Il est important de noter que l’expression de PARP1, l’enzyme de première ligne de la signalisation SSB, diminue également en réponse au stress oxydatif. Par conséquent, les « blocs » de SSB restent non réparés et s’accumulent. Cette accumulation active la p38MAPK qui induit la régulation à la hausse de l’inhibiteur du cycle cellulaire p16 et l’arrêt du cycle cellulaire. Contre-intuitivement, nous avons montré que l’accumulation des SSB non réparés est également responsable de la PSNE. En effet, la PSNE est abrogée par les anti-oxydants et par la ré-expression de PARP1 (Nassour et al, Nature Commun, 2016).
Par conséquent, une caractéristique récurrente majeure de la sénescence est l’accumulation de dommages non réparés à l’ADN. Cependant, selon la nature des dommages, le résultat cellulaire est très différent. Lorsque les dommages induisent la voie DDR, l’état sénescent est très stable et se comporte comme un état suppresseur de tumeur cellulaire autonome. En revanche, lorsque les dommages n’activent pas la voie DDR mais conduisent à l’arrêt du cycle cellulaire uniquement par p16 et lorsqu’ils sont associés à d’autres dommages organites d’origine oxydative, les cellules sénescentes ont deux conséquences: presque toutes meurent par autophagie, et quelques-unes échappent à la mort cellulaire autophagique et réintègrent le cycle cellulaire. En raison de la présence de SSB non réparés, les cellules filles acquièrent des mutations et des propriétés néoplasiques. Un tel mécanisme de sénescence est donc oncogène. Surtout, nous avons mis en évidence ces deux types de sénescence dans des coupes tissulaires de peau humaine, la première dans les fibroblastes du derme, la seconde dans les kératinocytes de l’épiderme (Nassour et al, Nature Commun, 2016).
Nous avons écrit trois articles de synthèse argumentant sur les mécanismes et les spécificités de la sénescence dans les cellules épithéliales (Nassour et Abbadie, Oncologie moléculaire et cellulaire, 2016; Abbadie et al, Cellulaire et moléculaire Life Sciences, 2017; Goy et Abbadie, Medecine / Sciences, 2018).
Contact :
Corinne Abbadie, Corinne.abbadie@ibl.cnrs.fr
A.4- Le sécrétome des fibroblastes sénescents favorise les événements cancérigènes précoces de la peau
Un autre paradigme bien démontré dans le domaine de la sénescence est que, bien qu’il soit dans un état suppresseur de tumeur, un fibroblaste sénescent produit un sécrétome modifié – le phénotype sécrétoire associé à la sénescence (SASP) – qui a des effets inflammatoires paracrines et favorisant la tumeur sur des cellules épithéliales déjà pré-transformées, mais pas sur des cellules épithéliales normales. Nous avons profité de notre modèle de PSNE pour déterminer si le SASP pouvait également stimuler les phases très précoces de la cancérogenèse. Nous avons démontré qu’un milieu de culture conditionné par des NHDF sénescents était capable d’augmenter la fréquence PSNE et d’augmenter la transition épithélium-mésenchyme des NHEKs PS. Nous avons mis en évidence les métalloprotéinases matricielles 1 et 2 (MMP1 et MMP2) comme les principaux composants du SASP responsable de ces changements. Nous avons identifié le récepteur activé par la protéase 1 (PAR1) comme un récepteur spécifiquement surexprimé dans les PS NHEK, activé par MMP1 et MMP2 et transduisant les changements de transition épithélium-mésenchyme (Malaquin et al. Plos One 2013).
Contact :
Albin Pourtier, albin.pourtier@ibl.cnrs.fr
A.5- La sénescence cellulaire implique une voie intracrine de prostaglandine E2 dans les NHDFs
Les médiateurs lipidiques tels que les prostaglandines pourraient également être des composants du SASP protumorigène en plus des cytokines, des facteurs de croissance et des MMP déjà décrits. Nous avions montré il y a quelques années que la COX-2, l’enzyme limitante et inductible de la voie de biosynthèse des prostaglandines, participe à la sénescence des NHDF (Zdanov et al. Exp Cell Res 2007). Nous avons donc étudié plus en détail comment cette voie induit la sénescence. Nous avons montré que la PGE2, principale prostaglandine produite par COX-2, est capable d’induire à la fois le début et le maintien de la sénescence. Elle ne le fait que lorsqu’elle est importée à l’intérieur de la cellule par le transporteur PGE2/lactate, ce qui indique que la PGE2 agit sur la sénescence plus via le pool de récepteurs EP intracellulaires que via ceux localisés à la surface cellulaire. Le traitement avec des agonistes, des antagonistes et l’inhiition des récepteurs EP par ARN interférence a révélé que, parmi les EP, EP3 était le plus impliqué dans la transduction des effets intracrines de la PGE2 (Pluquet et al. BBA Lipids 2013).
Contact :
Olivier Pluquet, olivier.pluquet@ibl.cnrs.fr
A.6- La sénescence est associée au stress du réticulum endoplasmique et à l’activation de la réponse UPR (Unfolded Protein Response)
Puisque le réticulum endoplasmique (RE) est le premier organite de la voie sécrétoire, nous avons émis l’hypothèse que les changements dans le SASP pourraient être la conséquence d’un stress du RE se produisant à la sénescence. Nous avons montré que les NHDFs présentent une expansion du RE associée à une activation légère et chronique de la voie de la réponse UPR. Nous avons étudié plus précisément l’activation des trois bras de l’UPR, PERK, IRE1 et ATF6a, qui contribuent à rétablir l’homéostasie du réticulum par des effecteurs spécifiques. Nous avons montré que l’inhibition de la voie ATF6a, et seule cette voie, supprimait en partie certains marqueurs de sénescence, dont l’augmentation de certains composants du SASP (Druelle et al, Oncotarget, 2016).
Nous avons ensuite postulé que l’UPR pourrait fonctionner en sénescence par la voie COX-2 décrite ci-dessus, car la plupart des enzymes et des récepteurs de cette voie sont associés à la membrane ER. L’inhibition de l’ATF6a par ARN interférence dans les NHDFs pré-sénescents, mais pas de PERK ou IRE1, a induit une diminution des expressions COX-2 et PGES1 et 2 ainsi qu’une diminution de la production de PGE2. Corrélativement, cela a retardé partiellement l’apparition de marqueurs de sénescence, y compris certains composants SASP. Ces effets ont été évités en favorisant l’importation de PGE2, mais pas seulement en fournissant de la PGE2 extracellulaire. Inversement, le traitement de NHDFs jeunes par des inducteurs de stress du RE tel que le dithiothréitol induit une sénescence prématurée accompagnée d’une surexpression de COX-2 et d’une production de PGE2. L’inhibition de l’activité COX-2 par NS398 bloque partiellement le phénotype sénescent prématuré induit par le dithiotreitol. Pris ensemble, ces résultats indiquent que le stress du RE est un inducteur de sénescence, opérant, au moins en partie, par une voie ATF6a / COX-2 / PGE2 / intracellulaire EP3 (Cormenier et al, Mechanisms of Aging and Development, 2018). Les rôles du stress du RE et de l’activation de l’UPR à la sénescence a été décrit dans Pluquet et al, Am J Physiol Cell Physiol, 2015.
Contact :
Olivier Pluquet, olivier.pluquet@ibl.cnrs.fr
Publications : voir liste de publications
Collaborations :
Local :
- Eric Adrieaensens (Canther)
- Sylviane Pied (CIIL, Institut Pasteur de Lille)
- Rejane Paumelle (EGID, UMR1011)
- Natacha Prevarskaya (U1003)
- Florence Renaud (CBP, CHRU Lille)
- Frank Lafont (CIIL, Institut Pasteur de Lille)
- Fabrizio Cleri, a Physicist at the IEMN
- Guillemette Marot, INRIA
National :
- Antoine Galmiche (EA4666, CHU of Amiens)
- Eric Chevet (ER440, Rennes)
- Olivier Coqueret (CRCINA, U1232, Angers)
International :
- Véronique Fontaine, Université Libre de Brussels (Belgium)
- Kevin Braekmans, a Physicist at the Gent University (Belgium)
- Florence Debacq-Chainiaux, Université de Namur (Belgium)
Financements :
- Université de Lille
- Région Nord-Pas-de-Calais
- SFR Cancer, Lille
- Cancéropole Nord-Ouest
- FEDER/Région Hauts de France/MEL/Etat/ Convention CTRL
- SIRIC ONCOLille
- Groupement des Entreprises Françaises dans la Lutte contre le Cancer (GEFLUC)
- Ligue contre le Cancer (comité du septentrion)
B- Induction et échappement à la sénescence des cellules cancéreuses coliques en réponse à la chimiothérapie
De nombreuses études ont montré que la sénescence peut être induite dans des cellules cancéreuses par nombre des agents génotoxiques utilisés en thérapie anti-néoplasique ; cette sénescence est appelée « sénescence induite par la thérapie » ou TIS (Therapy-Induced Senescence). Si la possibilité d’utiliser de telles thérapies pro-sénescence (qui appellent des doses plus faibles que les protocoles de référence pro-apoptose) pour le traitement du cancer colorectal (CCR) suscite un intérêt considérable, des études ont rapporté l’existence de mécanismes d’échappement à la TIS au cours desquels certaines cellules cancéreuses sénescentes sont capables de ré-entrer dans le cycle cellulaire, ce qui contribue probablement à la récidive tumorale.
Notamment, les cellules qui échappent à la TIS réacquièrent des caractéristiques de cellules souches et un phénotype d’autant plus transformé, limitant ainsi l’utilisation des thérapies pro-sénescence. Néanmoins, la découverte récente de molécules dites « sénolytiques » ouvre la possibilité de lever cette limite en éliminant les cellules cancéreuses entrées en TIS. Les molécules sénolytiques identifiées jusqu’à présent agissent principalement en ciblant certaines voies anti-apoptotiques up-régulées dans les cellules sénescentes, ouvrant ainsi la voie à de nouvelles stratégies thérapeutiques combinant TIS et sénolytiques. Mais avant de pouvoir envisager de telles stratégies combinatoires, une meilleure compréhension des mécanismes moléculaires régissant l’établissement et l’échappement à la TIS des cellules cancéreuses coliques apparait nécessaire.
Dans ce contexte, nous travaillons dans l’équipe, depuis plusieurs années, sur la voie de biosynthèse des hexosamines (HBP), une voie métabolique qui utilise le glucose extracellulaire et la glutamine pour synthétiser l’UDP-GlcNAc, substrat de la O-GlcNAc Transférase (OGT). Cette enzyme assure le transfert d’un résidu de GlcNAc sur des Sérines ou Thréonines de ses protéines cibles et est la seule glycosyltransférase capable de modifier post-traductionnellement des protéines cytosoliques et nucléaires. La O-GlcNAcylation des protéines nucléocytoplasmiques est réversible : la O-GlcNAcase (OGA) assure l’hydrolyse du résidu de GlcNAc.
Nos précédents travaux ont mis en évidence une augmentation des niveaux de O-GlcNAcylation et d’expression de l’OGT dans des échantillons de CCR humain par rapport aux tissus normaux (Olivier-Van Stichelen *, Dehennaut * et al., 2014), ainsi que dans un modèle murin de carcinogenèse colique (Decourcelle et al., 2020). De plus, nous avons montré que le « knock-down » de l’OGT par siRNA diminuait la prolifération, la survie et l’adhérence de cellules cancéreuses coliques (Steenackers et al., 2016). Ces travaux, parmi d’autres, ont permis de définir l’hyper-O-GlcNAcylation comme nouveau marqueur des CCR et comme une cible thérapeutique potentielle. En revanche, seules 4 études se sont penchées à ce jour sur le rôle potentiel de la O-GlcNAcylation dans la régulation de la sénescence. Ces études menées dans un contexte de sénescence induite par un stress oncogénique (OIS, Oncogene-Induced Senescence) ou par radiothérapie soutiennent l’hypothèse d’un effet inhibiteur de la voie HBP et de la O-GlcNAcylation sur ces 2 types de sénescence. Néanmoins, aucune étude ne s’était pour l’instant intéressée à l’implication de la voie HBP et de la O-GlcNAcylation dans le processus d’entrée et d’échappement à la sénescence induite par la chimiothérapie, c’est donc ce que nous avons récemment entrepris d’étudier. Nous avons d’ores et déjà obtenus des résultats préliminaires montrant une implication de la voie HBP et de la O-GlcNAcylation dans l’entrée en TIS de plusieurs lignées cancéreuses coliques et que nous sommes en train de conforter.
Contact : Vanessa Dehennaut, vanessa.dehennaut@ibl.cnrs.fr
Publications : voir liste de publications
Collaborations :
Local :
- A. Vincent, N. Jonckheere (CANTHER UMR1277-U9020).
- Pr I Belkoura-El Yazidi ; Dr Anne-Sophie Vercoutter-Edouart (Unité de Glycobiologie Structurale et Fonctionnelle, CNRS UMR 8576).
- Dr G. Certad ; Dr S. Benamrouz (Centre d’Infection et d’Immunité de Lille, INSERM U1019, CNRS UMR 8204).
International :
Dr A. Escobar-Ramirez (Université autonome de Tabasco, Mexique).
Financements :
- Cancéropole Nord-Ouest
- Ligue contre le Cancer, Comité du septentrion
- GEFLUC Flandres Artois
C- ARN non codants dans le cancer et pendant la fibrogenèse
C.1- Comprendre le rôle des miARN pendant la fibrogénèse.
La fibrose est la dernière voie commune dans pratiquement toutes les formes d’insuffisance organique chronique, y compris les poumons, le foie et les reins, et est une des principales causes de morbidité et de mortalité dans le monde. La fibrose résulte de l’activité excessive des fibroblastes, en particulier une forme différenciée connue sous le nom de myofibroblaste qui est responsable de l’accumulation excessive et persistante de tissu cicatriciel et, finalement, d’une défaillance organique. Comme les études mécanistes ont lié les ARNc à un large éventail de troubles humains complexes, les thérapies basées sur l’ARNc offrent de nouvelles opportunités formidables pour traiter les maladies incurables. Dans cette optique, nous avons estimé que les ARNnc jouent un rôle important dans les événements pathogènes conduisant à la fibrogénèse et peuvent donc représenter de nouveaux biomarqueurs diagnostiques / pronostiques ainsi que de nouvelles cibles médicamenteuses précieuses.
Le processus de fibrogenèse (rénale, pulmonaire et hépatique) a jusqu’à présent été appréhendé dans plusieurs contextes pathologiques (néphropathie à IgA, utilisation d’immunosuppresseurs chroniques, exposition aux métaux lourds, fibrose pulmonaire idiopathique), en utilisant des modèles cellulaires (lignées cellulaires épithéliales et fibroblastiques, cultures primaires) des cellules épithéliales), des modèles murins (obstruction urétérale unilatérale, ischémie-reperfusion rénale, pompes osmotiques délivrant des immunosuppresseurs, induction de la fibrose pulmonaire par instillation de bléomycine, induction de la fibrose hépatique par ligature des voies biliaires ou injections CCl4 …) et cohortes de patients. Des analyses transcriptomiques à haut débit nous ont permis de montrer que certains miARN sont couramment impliqués dans le développement de la fibrose pulmonaire, rénale et hépatique.
a / En particulier, nous avons caractérisé l’implication de trois miARN (miR-199a-5p (brevet déposé), miR-199a-3p et miR-214-3p) générés à partir du même locus, LncRNA DNM3OS, un long ARN non codant, (Lino Cardenas et al., 2013, Savary et al., 2019). Les miARN de ce cluster participent à l’activation des fibroblastes et à leur différenciation en myofibroblastes via la régulation des voies canoniques et non canoniques du TGFβ, ciblant notamment CAV1 (miR-199a-5p) et GS3Kβ (miR-214-3p). Nous avons également montré que DNM3OS est également impliqué dans les mécanismes de réparation épithéliale en régulant les facteurs KGF et HGF (miR-199a-3p). Enfin, nous avons montré que l’interférence avec la fonction DNM3OS en utilisant des stratégies pharmacologiques distinctes (anti-miR, Target Site Blocker et gapmer) prévient non seulement la fibrose pulmonaire, hépatique et rénale, mais améliore également la fibrose pulmonaire établie, fournissant ainsi un nouveau paradigme pour le traitement des maladies fibrotiques (Savary et al., 2019).
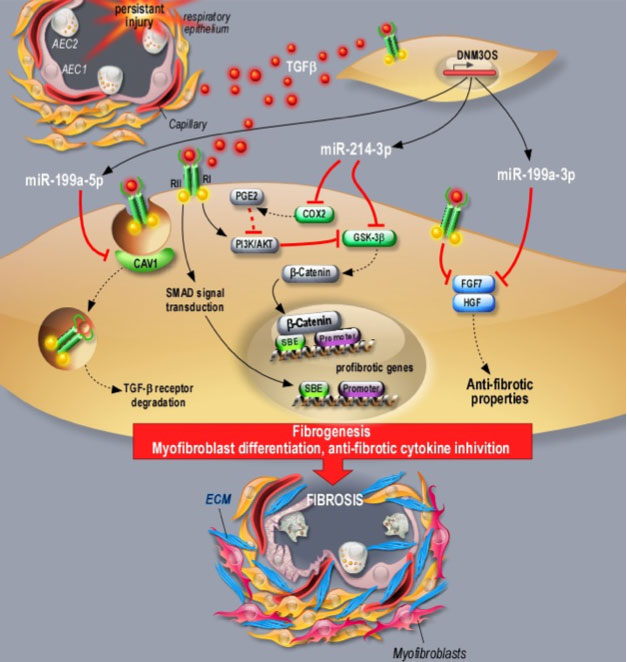
b / Par ailleurs, nous avons confirmé le rôle du miR-21 dans le processus de fibrose rénale (Glowacki et al., 2013, Hennino et al., 2016) puis proposé l’intérêt de ce miRNA comme biomarqueur non invasif de la fibrose rénale, les taux sériques de miR-21 étant corrélés à la gravité des lésions de fibrose rénale (Glowacki et al., 2013). Le rôle prépondérant du miR-21 a également été évalué dans le contexte de la néphrotoxicité induite par des produits chimiques, en particulier le cadmium (Lemaire et al., 2020) et l’utilisation chronique de l’inhibiteur de la calcineurine (Vandenbussche et al., 2018).
Contact :
Pottier Nicolas, MCU-PH, nicolas.pottier@univ-lille.fr
Cauffiez Christelle, MCU, christelle.cauffiez@univ-lille.fr
C.2- Identification de nouveaux déterminants moléculaires sous-jacents à la réponse au cisplatine en terme d’efficacité et de néphrotoxicité
Le cisplatine, un médicament chimiothérapeutique de première intention largement utilisé pour divers cancers, a attiré une attention soutenue de la recherche depuis son introduction sur le marché il y a plus de 40 ans. Bien que le cisplatine rétrécisse efficacement la tumeur, son accumulation non spécifique à la fois dans la tumeur et dans les tissus normaux a également provoqué des effets secondaires graves tels que la néphrotoxicité. Par conséquent, des efforts de recherche intensifs sont consacrés à l’amélioration de l’efficacité antitumorale du cisplatine tout en améliorant son profil d’innocuité. Dans ce contexte, nous avons développé deux projets, l’un visant à découvrir de nouveaux déterminants moléculaires médiant la résistance au cisplatine dans le cancer du poumon et l’autre lié à la prévention de la néphrotoxicité induite par le cisplatine.
2.a: Identification de nouveaux déterminants moléculaires sous-jacents à la résistance au cisplatine.
Nous avons effectué un criblage fonctionnel de la bibliothèque de miARN pour identifier les miARN associés à la résistance au cisplatine. Le dépistage effectué avec environ 1000 mimétiques a révélé que la surexpression de miR-24-3p, un miARN identifié comme le meilleur hit, induit de manière reproductible la résistance au cisplatine des cellules cancéreuses du poumon A549. Enfin, nous avons pu démontrer à travers de multiples approches in vitro indépendantes que le miR-24-3p est un puissant régulateur non seulement de la mort des cellules pulmonaires induite par le cisplatine, mais également de la réponse aux thérapies ciblées par l’EGFR (brevet déposé).
Contact: Pottier Nicolas, MCU-PH, nicolas.pottier@univ-lille.fr
2.b: Identification de nouveaux déterminants moléculaires sous-jacents à la néphrotoxicité du cisplatine. Dans le but de découvrir de nouvelles approches pharmacologiques prévenant les effets néphrotoxiques du cisplatine, nous avons cherché à tester deux stratégies potentielles. Nous avons d’abord émis l’hypothèse que l’inhibition du miR-21, qui est principalement impliqué dans les lésions cellulaires, pourrait diminuer les lésions rénales de cisplatine. Nos premiers résultats ont indiqué que le miR-21 joue un rôle ambivalent dans les lésions rénales et semble être protecteur à un stade précoce, ou délétère lorsque le processus se prolonge dans le temps. De plus, nous avons initié une étude prospective visant à découvrir de nouveaux paramètres cliniques corrélés à la néphrotoxicité induite par le cisplatine (collaboration avec le Département d’Oncologie Thoracique, CHRU, Lille).
Contact: Cauffiez Christelle, MCU, christelle.cauffiez@univ-lille.fr
Publications : voir liste de publications
Collaborations :
Local :
- Dr Blum (INSERM U1172, JPARC)
- Dr Perrais (Canther)
- Pr Boulanger (LIRIC UMR U995)
- Dr Gnemmi et Dr Gibier (Institut de pathologie, CHU Lille)
- Pr Hazzan & Dr Lionet (Service de Néphrologie, CHU Lille)
National :
- Dr Barbry & Dr Mari (CNRS IMPC, Sophia-Antipolis, Valbonne)
- Pr Marquette (Service de Pneumologie, CHRU Nice)
- Pr Hertig (Urgences Néphrologiques et Transplantation Rénale, Hôpital Tenon Paris)
International :
- Dr Luedde & Dr Roderburg (Department of Medicine III, University Hospital RWTH, Aachen, Germany)
- Dr Kaminski (Pulmonary, Critical Care and Sleep Medicine, Yale School of Medicine, New Heaven, CT, USA)
- Dr Laumet (Department of Physiology, University of Michigan)
Financements :
- ANR (FibromiR)
- SIRIC ONCOLille
- SATT Nord
- Santélys
- Ligue contre le cancer
- Astellas
- Novartis
- Plan Cancer
- FEDER/Région Hauts de France/MEL/Etat/ Convention CTRL
- Région Nord-Pas-de-Calais
- Cancéropôle Nord Ouest
> ARTICLES ORIGINAUX DU LABORATOIRE
> REVUES GÉNÉRALES
> LETTRES – COMMENTAIRES
> ÉDITORIAL
> CHAPITRES DE LIVRE
> ARTICLES ORIGINAUX ET REVUES GÉNÉRALES ISSUS DES COLLABORATIONS
> PUBLICATIONS CLINIQUES
Publications 2023-2015
ARTICLES ORIGINAUX DU LABORATOIRE
2023
Lionet L, Van Triempon M, Figeac M, Fages V, Gibier JB, Provot F, Maanaoui M, Pottier N, Cauffiez C, Glowacki F. Extracorporeal photopheresis reduces fibrotic and inflammatory transcriptomic biological marker of chronic antibody-mediated kidney rejection. Transplant Direct, in press
Larrue R, Fellah S, Boukrout N, De Sousa C, Lemaire J, Leboeuf C, Goujon M, Perrais M, Mari B, Cauffiez C, Pottier N*, Van der Hauwaert C*. miR-92a-3p regulates cisplatin-induced cancer cell death. Cell Death Dis. 2023 Sep 13;14(9):603. doi: 10.1038/s41419-023-06125-z.
Goy E, Martin N, Drullion C, Saas L, Molendi-Coste O, Pineau L, Dombrowicz D, Deruy E, Bauderlique-Le-Roy H, Samyn O, De Launoit Y, Abbadie C. Flow Cytometry- based Method for Efficient Sorting of Senescent Cells. Bio Protoc. 2023 Apr 5;13(7):e4612. doi: 10.21769/BioProtoc.4612.
Mound A, Goormachtigh G, Bray F, Flament S, Rolando C, Ruez R, Martin N, Decourcelle A, Dehennaut V, Saliou JM, Chamaillard M, Abbadie C. The NLRP6 protein is very faintly expressed in several normal and cancerous epithelial cells and may be confused with an unrelated protein. PLoS One. 2023 Jan 20;18(1):e0279028. doi: 10.1371/journal.pone.0279028. PMID: 36662875; PMCID: PMC9858803. IF=3.752
2022
Dewaeles E, Carvalho K, Fellah S, Sim J, Boukrout N, Caillierez R, Ramakrishnan H, Van der Hauwaert C, Vijaya Shankara J, Martin N, Massri N, Launay A, Folger JK, de Schutter C, Larrue R, Loison I, Goujon M, Jung M, Le Gras S, Gomez-Murcia V, Faivre E, Lemaire J, Garat A, Beauval N, Maboudou P, Gnemmi V, Gibier JB, Buée L, Abbadie C, Glowacki F, Pottier N, Perrais M, Cunha RA, Annicotte JS, Laumet G, Blum D, Cauffiez C. Istradefylline protects from cisplatin-induced nephrotoxicity and peripheral neuropathy while preserving cisplatin antitumor effects. J Clin Invest. 2022 Nov 15;132(22):e152924. doi: 10.1172/JCI152924. PMID: 36377661; PMCID: PMC9663157. IF=19.456
Goy E, Tomezak M, Facchin C, Martin N, Bouchaert E, Benoit J, de Schutter C, Nassour J, Saas L, Drullion C, Brodin PM, Vandeputte A, Molendi-Coste O, Pineau L, Goormachtigh G, Pluquet O, Pourtier A, Cleri F, Lartigau E, Penel N, Abbadie C. The out-of-field dose in radiation therapy induces delayed tumorigenesis by senescence evasion. Elife. 2022 Mar 18;11:e67190. doi: 10.7554/eLife.67190. PMID: 35302491; PMCID: PMC8933005. IF=8.713
Larrue R, Fellah S, Van der Hauwaert C, Hennino MF, Perrais M, Lionet A, Glowacki F, Pottier N, Cauffiez C. The Versatile Role of miR-21 in Renal Homeostasis and Diseases. Cells. 2022 Nov 7;11(21):3525. doi: 10.3390/cells11213525. PMID: 36359921; PMCID: PMC9657972. IF=7.666Fellah S, Larrue R, Truchi M, Vassaux G, Mari B, Cauffiez C, Pottier N. Pervasive role of the long noncoding RNA DNM3OS in development and diseases. Wiley Interdiscip Rev RNA. 2022 May 1:e1736. doi: 10.1002/wrna.1736. Epub ahead of print. PMID: 35491542. IF=9.349
Goujon M, Woszczyk J, Gaudelot K, Swierczewski T, Fellah S, Gibier JB, Van Seuningen I, Larrue R, Cauffiez C, Gnemmi V, Aubert S, Pottier N, Perrais M. A Double-Negative Feedback Interaction between miR-21 and PPAR-α in Clear Renal Cell Carcinoma. Cancers (Basel). 2022 Feb 4;14(3):795. doi: 10.3390/cancers14030795. PMID: 35159062; PMCID: PMC8834244. IF=6.575
2021
Paget S, Dubuissez M, Page A, Dehennaut V, Loison I, Spruyt N, Leprince D. Phosphorylation of HIC1 (Hypermethylated in Cancer 1) Ser694 by ATM is essential for DNA repair. Biochem Biophys Res Commun. 2021 Mar 20;553:51-57. doi:10.1016/j.bbrc.2021.03.060. Epub ahead of print. PMID: 33756345. IF: 2.985Gibier JB, Swierczewski T, Csanyi M, Hemon B, Glowacki F, Maboudou P, Van Seuningen I, Cauffiez C, Pottier N, Aubert S, Perrais M, Gnemmi V. MUC1 Mitigates Renal Injury and Inflammation in Endotoxin Induced Acute Kidney Injury by Inhibiting the TLR4-MD2 Axis and Reducing Pro-Inflammatory Macrophages Infiltration. Shock. 2021 Jan 28. doi: 10.1097/SHK.0000000000001742. Epub ahead of print. PMID: 33534395. IF: 2.960
Laboux T, Gibier JB, Pottier N, Glowacki F, Hamroun A. COVID-19-related collapsing glomerulopathy revealing a rare risk variant of APOL1: lessons for the clinical nephrologist. J Nephrol. 2021 Feb 6:1–6. doi: 10.1007/s40620-020-00935-6. Epub ahead of print. Erratum in: J Nephrol. 2021 Apr 3;: PMID: 33548053; PMCID: PMC7865108. IF : 9.274
Hamroun A, Camier A, Bigna JJ, Glowacki F. Impact of air pollution on renal outcomes: a systematic review and meta-analysis protocol. BMJ Open. 2021 Jan 17;11(1):e041088. doi: 10.1136/bmjopen-2020-041088. PMID: 33455930; PMCID: PMC7813312. IF : 2.496
Gnemmi V, Gibier JB, Humez S, Copin MC, Glowacki F. Néphrite interstitielle granulomateuse : le point de vue du pathologiste [Renal granulomatous nephritis: Histopathological point of view]. Ann Pathol. 2021 Apr;41(2):166-175. French. doi: 10.1016/j.annpat.2020.11.001. Epub 2020 Dec 1. PMID: 33277052. IF : 0.394
Paccou J, Pflimlin A, Glowacki F, Cortet B. A Challenging Case of Tumor-Induced Osteomalacia. Am J Med. 2021 Jan;134(1):e60-e61. doi: 10.1016/j.amjmed.2020.06.032. Epub 2020 Jul 24. PMID: 32712146. IF : 4.760
2020
Lemaire J, Larrue R, Perrais M, Cauffiez C, Pottier N. Aspects fondamentaux du développement tumoral [Fundamental aspects of oncogenesis]. Bull Cancer. 2020 Nov;107(11):1148-1160. French. doi: 10.1016/j.bulcan.2020.08.004. Epub 2020 Oct 7. PMID: 33039132. IF : 0.912
Larrue R, Chamley P, Bardyn T, Lionet A, Gnemmi V, Cauffiez C, Glowacki F, Pottier N, Broly F. Diagnostic utility of whole-genome sequencing for nephronophthisis. NPJ Genom Med. 2020 Sep 21;5:38. doi: 10.1038/s41525-020-00147-8. PMID: 33024573; PMCID: PMC7506526. IF : 5.631
Dubuissez M, Paget S, Abdelfettah S, Spruyt N, Dehennaut V, Boulay G, Loison I, de Schutter C, Rood BR, Duterque-Coquillaud M, Leroy X, Leprince D. HIC1 (Hypermethylated in Cancer 1) modulates the contractile activity of prostate stromal fibroblasts and directly regulates <i>CXCL12</i> expression. Oncotarget. 2020 Nov 10;11(45):4138-4154. doi: 10.18632/oncotarget.27786. PMID: 33227080; PMCID: PMC7665237. IF: 3.765
Abdelfettah S, Boulay G, Dubuissez M, Spruyt N, Garcia SP, Rengarajan S, Loison I, Leroy X, Rivera MN, Leprince D. hPCL3S promotes proliferation and migration of androgen-independent prostate cancer cells. Oncotarget. 2020 Mar 24;11(12):1051-1074. doi: 10.18632/oncotarget.27511 IF: 3.71
Abbadie C, Pluquet O. Unfolded Protein Response (UPR) Controls Major Senescence Hallmarks. Trends in Biochemical Sciences, 2020, 45(5):371-374. IF: 16.88
Perry A, Douillard C, Jonca F, Glowacki F, Leroy X, Caveriviere P, Hubert A, Labrune P. Papillary renal cell carcinoma in two young adults with glycogen storage disease type Ia. JIMD Rep. 2020 Jan 29;52(1):17-22. doi: 10.1002/jmd2.12096. IF: 4.287
Delsart P, Vambergue A, Ninni S, Machuron F, Lelievre B, Ledieu G, Fontaine P, Merlen E, Frimat M, Glowacki F, Montaigne D, Mounier-Vehier C. Prognostic significance of the renal resistive index in the primary prevention of type II diabetes. J Clin Hypertens (Greenwich). 2020 Feb;22(2):223-230. doi: 10.1111/jch.13819. IF: 2.444
Bitton L, Vandenbussche C, Wayolle N, Gibier JB, Cordonnier C, Verine J, Humez S, Bataille P, Lenain R, Ramdane N, Azar R, Mac Namara E, Hatron PY, Maurage CA, Perrais M, Frimat M, Vanhille P, Glowacki F, Buob D, Copin MC, Quéméneur T, Gnemmi V. Tubulointerstitial damage and interstitial immune cell phenotypes are useful predictors for renal survival and relapse in antineutrophil cytoplasmic antibody-associated vasculitis. J Nephrol. 2020 Jan 8. doi: 10.1007/s40620-019-00695-y. IF: 3.698
Lemaire J, Van der Hauwaert C, Savary G, Dewaeles E, Perrais M, Lo Guidice JM, Pottier N, Glowacki F, Cauffiez C. Cadmium-Induced Renal Cell Toxicity Is Associated With MicroRNA Deregulation. Int J Toxicol. 2020 Mar/Apr;39(2):103-114.doi: 10.1177/1091581819899039. IF: 1.223
Decourcelle A, Loison I, Baldini S, Leprince D, Dehennaut V. Evidence of a compensatory regulation of colonic O-GlcNAc transferase and O-GlcNAcase expression in response to disruption of O-GlcNAc homeostasis. Biochem Biophys Res Commun. 2020 Jan 1;521(1):125-130. doi: 10.1016/j.bbrc.2019.10.090. IF: 2.705
Decourcelle A, Very N, Djouina M, Loison I, Thévenet J, Body-Malapel M, Lelièvre E, Coqueret O, Leprince D, El Yazidi-Belkoura I, Dehennaut V. O-GlcNAcylation Links Nutrition to the Epigenetic Downregulation of UNC5A during Colon Carcinogenesis. Cancers (Basel). 2020 Oct 28;12(11):3168. doi: 10.3390/cancers12113168. PMID: 33126652; PMCID: PMC7693889. IF : 5.968
Pekar JD, Grzych G, Durand G, Haas J, Lionet A, Brousseau T, Glowacki F, Maboudou P. Calcium state estimation by total calcium: the evidence to end the never-ending story. Clin Chem Lab Med. 2020 Jan 28;58(2):222-231. doi: 10.1515/cclm-2019-0568. IF: 3.638
2019
Savary G, Dewaeles E, Diazzi S, Buscot M, Nottet N, Fassy J, Courcot E, Henaoui I-S, Lemaire J, Martis N, Van der Hauwaert C, Pons N, Magnone V, Leroy S, Plantier L, Lebrigand K, Paquet A, Lino Cardenas CL, Vassaux G, Crestani B, Wallaert B, Rezzonico R, Brousseau T, Glowacki F, Bellusci S, Perrais M, Broly F, Barbry P, Marquette C-H, Cauffiez C, Mari B, Pottier N. The long non-coding RNA DNM3OS is a reservoir of fibromiRs with major functions in lung fibroblast response to TGF-β and pulmonary fibrosis. Am J Respir Crit Care Med. 2019 Jul 15;200(2):184-198.
Maanaoui M, Lenain R, Hamroun A, Van der Hauwaert C, Lopez B, Gibier JB, Frimat M, Savary G, Hennart B, Larrue R, Pottier N, Broly F, Provôt F, Hazzan M, Glowacki F, Cauffiez C. Caveolin-1 rs4730751 single-nucleotide polymorphism may not influence kidney transplant allograft survival. Sci Rep. 2019 Oct 29;9(1):15541.
2018
Drullion C, Marot G, Martin N, Deslé J, Saas L, Salazar-Cardozo C, Bouali F, Pourtier A, Abbadie C and Pluquet O. (2018) Pre-malignant transformation by senescence evasion is prevented by the PERK and ATF6alpha branches of the Unfolded Protein Response. Cancer Letters. 438 :187-196
Cormenier J, Martin N, Deslé J, Salazar-Cardozo C, Pourtier A, Abbadie C, Pluquet O. The ATF6α arm of the Unfolded Protein Response mediates replicative senescence in human fibroblasts through a COX2/prostaglandin E(2) intracrine pathway. Mech Ageing Dev. 2018, 170, 82-91
Vandenbussche C*, Van der Hauwaert C*, Dewaeles E, Franczak J, Hennino MF, Gnemmi V, Savary G, Tavernier Q, Nottet N, Paquet A, Perrais M, Blum D, Mari B, Pottier N, Glowacki F, Cauffiez C. Tacrolimus-induced nephrotoxicity in mice is associated with microRNA deregulation. Arch Toxicol. 2018, 92, 1539-1550
2017
Paget S, Dubuissez M, Dehennaut V, Nassour J, Harmon BT, Spruyt N, Loison I, Abbadie C, Rood BR, Leprince D. (2017) HIC1 (hypermethylated in cancer 1) SUMOylation is dispensable for DNA repair but is essential for the apoptotic DNA damage response (DDR) to irreparable DNA double-strand breaks (DSBs). Oncotarget 10;8(2):2916-2935.
2016
Tomezak, C. Abbadie, E. Lartigau and F. Cleri. A biophysical model of cell evolution after cytotoxic treatments: damage, repair and cell response. Journal of Theoretical Biology, 2016, 389, 146-58
Nassour, S. Martien, E. Deruy, E. Tomellini, N. Malaquin, N. Martin, F. Bouali, L. Sabatier, N. Wernert, S. Pinte, E. Gilson, A. Pourtier, O. Pluquet, C. Abbadie. Defective DNA single-strand break repair is responsible for senescence and neoplastic escape of epithelial cells. Nature Communications, 2016, 7:10399
Druelle*, C. Drullion*, J. Deslé*, N. Martin, L. Saas, J. Cormenier, N. Malaquin, L. Huot, C. Slomianny, F. Bouali, C. Vercamer, D. Hot, A. Pourtier, E. Chevet, C. Abbadie, O. Pluquet. ATF6a regulates morphological changes associated with senescence in human fibroblasts. Oncotarget, 2016, 7, 67699-67715
Hennino MF, Buob D, Van der Hauwaert C, Gnemmi V, Jomaa Z, Pottier N, Savary G, Drumez E, Noël C, Cauffiez C, Glowacki F. miR-21-5p renal expression is associated with fibrosis and renal survival in patients with IgA nephropathy. Sci Rep. 2016, 6, 27209
2015
Van der Hauwaert C, Savary G, Pinçon C, Gnemmi V, Noël C, Broly F, Labalette M, Perrais M, Pottier N, Glowacki F, Cauffiez C. Donor caveolin 1 (CAV1) genetic polymorphism influences graft function after renal transplantation. Fibrogenesis Tissue Repair. 2015, 5, 8.
REVUES GÉNÉRALES
2023
Giroud J, Bouriez I, Paulus H, Pourtier A, Debacq-Chainiaux F, Pluquet O. Exploring the Communication of the SASP: Dynamic, Interactive, and Adaptive Effects on the Microenvironment. Int J Mol Sci. 2023 Jun 28;24(13):10788. doi: 10.3390/ijms241310788.
Crespin T, Lionet A, Augusto JF, Hazzan M, Garrouste C, Heng AE, Ducloux D. Immunomodulation par photo-chimiothérapie extra corporelle en transplantation rénale : preuves de concept, données cliniques et perspectives.NET-2023-23/R2; in press
Fellah S, Larrue R, Truchi M, Vassaux G, Mari B, Cauffiez C, Pottier N. Pervasive role of the long noncoding RNA DNM3OS in development and diseases. Wiley Interdiscip Rev RNA. 2023 Mar;14(2):e1736. doi: 10.1002/wrna.1736.
Hamroun A, Glowacki F. Le traitement conservateur dans la prise en charge de la maladie rénale chronique [Comprehensive conservative care for the management of advanced chronic kidney disease]. Nephrol Ther. 2023 Jun 29;19(S1):21-29.
Hamroun A, Glowacki F, Frimat L. Comprehensive Conservative Care: what doctors say, what patients hear. Nephrol Dial Transplant. 2023 May 8:gfad088. doi: 10.1093/ndt/gfad088
2022
Larrue R, Fellah S, Van der Hauwaert C, Hennino MF, Perrais M, Lionet A, Glowacki F, Pottier N, Cauffiez C. The Versatile Role of miR-21 in Renal Homeostasis and Diseases. Cells. 2022 Nov 7;11(21):3525. doi: 10.3390/cells11213525.
Lionet A, Urena Torres PA. La calciphylaxie urémique [Uremic calciphylaxis]. Nephrol Ther. 2022 Jun;18(3):180-188. French.
doi: 10.1016/j.nephro.2021.12.005.
2021
Pluquet O. and Abbadie C, Cellular senescence and tumor promotion: Role of the Unfolded Protein Response. Advances in Cancer Research. 2021 in press.
2020
Abbadie C, Pluquet O. Unfolded Protein Response (UPR) Controls Major Senescence Hallmarks. Trends in Biochemical Sciences, 2020 in press. IF: 16.88
2019
Pluquet O, and Galmiche A. (2019) Impact and relevance of the Unfolded Protein Response in HNSCC. Int J Mol Sci. 20. pii:E2654
Van der Hauwaert C, Glowacki F, Pottier N, Cauffiez C. Non-Coding RNAs as New Therapeutic Targets in the Context of Renal Fibrosis. Int J Mol Sci. 2019 Apr 23;20(8). pii: E1977.
Decourcelle A, Leprince D, Dehennaut V. (2019) Regulation of Polycomb repression by O-GlcNAcylation: linking nutrition to epigenetic reprogramming in embryonic development and cancer. Front Endocrinol (Lausanne) 10:117.
Pluquet O, Abbadie C, and Coqueret O. (2019) Connecting cancer relapse with senescence. Cancer Letters. 453: 50-58.
Hamroun A, Lenain R, Bigna JJ, Speyer E, Bui L, Chamley P, Pottier N, Cauffiez C, Dewaeles E, Dhalluin X, Scherpereel A, Hazzan M, Maanaoui M, Glowacki F. Prevention of Cisplatin-Induced Acute Kidney Injury: A Systematic Review and Meta-Analysis. Drugs 2019, Sep;79(14):1567-1582
2018
Goy and C. Abbadie. Senescence and cancer : double-dealing. Médecine/Sciences, 2018, 34,223-230
2017
Abbadie, O. Pluquet and A. Pourtier. Epithelial cell senescence : an adaptive response to pre-carcinogenic stresses. Cellular and Molecular Life Sciences, 2017, 74, 4471-4509
Galmiche A, Sauzay C, Chevet E and Pluquet O. Role of the Unfolded Protein Response in tumour cell characteristics and cancer outcome. Curr Opin Oncol. 2017, 29(1):41-47
2016
Nassour and C. Abbadie. A novel role for DNA single-strand breaks in senescence and neoplastic escape of epithelial cells. Molecular and Cellular Oncology, 2016, 3, e1190885
Galmiche A, Sauzay C, Houessinon A, chauffert B, and Pluquet O. Probing tumour proteostasis and the UPR with serum markers. Trends Cancer, 2016, 2:219-221.
2015
Pluquet, A. Pourtier and C. Abbadie. The Unfolded Protein Response and Cellular Senescence. Am J Physiol Cell Physiol, 2014, 308, C415-C425
Van der Hauwaert C, Savary G, Hennino MF, Pottier N, Glowacki F, Cauffiez C. [MicroRNAs in kidney fibrosis]. Nephrol Ther. 2015, 11, 474-482
Furlan A, Pourtier, A. Ets-1 activation, when tumors crosstalk with their microenvironment. Can Cell Microenviron. 2015. 2 (1). doi: 10.14800/ccm.494
LETTRES – COMMENTAIRES
2020
Lemaire J, Larrue R, Perrais M, Cauffiez C, Pottier N. Aspects fondamentaux du développement tumoral [Fundamental aspects of oncogenesis]. Bull Cancer. 2020 Nov;107(11):1148-1160. French. doi: 10.1016/j.bulcan.2020.08.004. Epub 2020 Oct 7. PMID: 33039132.
2019
Savary G, Pottier N, Mari B, Cauffiez C. The function of a long non coding RNA decoded in idiopathic pulmonary fibrosis. Med Sci (Paris). 2019 Oct;35(10):739-742.
ÉDITORIAL
—
CHAPITRES DE LIVRE
2017
Pluquet O, Pourtier A, et Abbadie C. La sénescence cellulaire dans les fibroblastes de derme : impact sur le vieillissement. In book: Biologie Cutanée « CoBip 2017″, Edition: MatriX, Chapter 7: 117-134, Publisher: SEMACO-COREP, Editor: Marek Haftek, ISSN 2266-9949
ARTICLES ORIGINAUX ET REVUES GÉNÉRALES ISSUS DES COLLABORATIONS
2023
Bauwens E, Parée T, Meurant S, Bouriez I, Hannart C, Wéra AC, Khelfi A, Fattaccioli A, Burteau S, Demazy C, Fransolet M, De Schutter C, Martin N, Théry J, Decanter G, Penel N, Bury M, Pluquet O, Garmyn M, Debacq-Chainiaux F. Senescence Induced by UVB in Keratinocytes Impairs Amino Acids Balance. J Invest Dermatol. 2023 Apr;143(4):554-565.e9. doi: 10.1016/j.jid.2022.11.017.
Altini N, Rossini M, Turkevi-Nagy S, Pesce F, Pontrelli P, Prencipe B, Berloco F, Seshan S, Gibier JB, Pedraza Dorado A, Bueno G, Peruzzi L, Rossi M, Eccher A, Li F, Koumpis A, Beyan O, Barratt J, Vo HQ, Mohan C, Nguyen HV, Cicalese PA, Ernst A, Gesualdo L, Bevilacqua V, Becker JU. Performance and limitations of a supervised deep learning approach for the histopathological Oxford Classification of glomeruli with IgA nephropathy. Comput Methods Programs Biomed. 2023 Sep 13;242:107814. doi: 10.1016/j.cmpb.2023.107814.
Martoriati A, Molinaro C, Marchand G, Fliniaux I, Marin M, Bodart JF, Takeda- Uchimura Y, Lefebvre T, Dehennaut V, Cailliau K. Follicular cells protect Xenopus oocyte from abnormal maturation via integrin signaling downregulation and O-GlcNAcylation control. J Biol Chem. 2023 Jun 23;299(8):104950. doi: 10.1016/j.jbc.2023.104950.
Isnard P, Vergnaud P, Garbay S, Jamme M, Eloudzeri M, Karras A, Anglicheau D, Galantine V, Jalal Eddine A, Gosset C, Pourcine F, Zarhrate M, Gibier JB, Rensen E, Pietropaoli S, Barba-Spaeth G, Duong-Van-Huyen JP, Molina TJ, Mueller F, Zimmer C, Pontoglio M, Terzi F, Rabant M. A specific molecular signature in SARS-CoV-2-infected kidney biopsies. JCI Insight. 2023 Mar 8;8(5):e165192. doi:
10.1172/jci.insight.165192.
Rensen E, Pietropaoli S, Mueller F, Weber C, Souquere S, Sommer S, Isnard P, Rabant M, Gibier JB, Terzi F, Simon-Loriere E, Rameix-Welti MA, Pierron G, Barba-Spaeth G, Zimmer C. Sensitive visualization of SARS-CoV-2 RNA with CoronaFISH. Life Sci Alliance. 2022 Jan 7;5(4):e202101124. doi: 10.26508/lsa.202101124.
Tusseau M, Lovšin E, Samaille C, Pescarmona R, Mathieu AL, Maggio MC, Selmanović V, Debeljak M, Dachy A, Novljan G, Janin A, Januel L, Gibier JB, Chopin E, Rouvet I, Goncalves D, Fabien N, Rice GI, Lesca G, Labalme A, Romagnani P, Walzer T, Viel S, Perret M, Crow YJ, Avčin T, Cimaz R, Belot A. DNASE1L3 deficiency, new phenotypes, and evidence for a transient type I IFN signaling. J Clin Immunol. 2022 Aug;42(6):1310-1320. doi: 10.1007/s10875-022-01287-5.
2022
Gonzales F, Guilmatre A, Barthélémy A, Lapillonne H, Pottier N, Leverger G, Petit A, Cheok MH. Ex vivo drug sensitivity profiling-guided treatment of a relapsed pediatric mixed-phenotype acute leukemia with venetoclax and azacitidine. Pediatr Blood Cancer. 2022
Oct;69(10):e29678. doi:10.1002/pbc.29678.
Mendez-Bermudez A, Lototska L, Pousse M, Tessier F, Croce O, Latrick CM, Cherdyntseva V, Nassour J, Xiaohua J, Lu Y, Abbadie
C, Gagos S, Ye J, Gilson E. Selective pericentromeric heterochromatin dismantling caused by TP53 activation during senescence. Nucleic Acids Res. 2022 Jul 22;50(13):7493-7510. doi: 10.1093/nar/gkac603.
Very N, Hardivillé S, Decourcelle A, Thévenet J, Djouina M, Page A, Vergoten G, Schulz C, Kerr-Conte J, Lefebvre T, Dehennaut V, El Yazidi-Belkoura I. Thymidylate synthase O-GlcNAcylation: a molecular mechanism of 5-FU sensitization in colorectal cancer. Oncogene. 2022 Jan;41(5):745-756. doi: 10.1038/s41388-021-02121-9
2021
Raab S, Gadault A, Very N, Decourcelle A, Baldini S, Schulz C, Mortuaire M, Lemaire Q, Hardivillé S, Dehennaut V, El Yazidi-Belkoura I, Vercoutter-Edouart AS, Panasyuk G, Lefebvre T. Dual regulation of fatty acid synthase (FASN) expression by O-GlcNAc transferase (OGT) and mTOR pathway in proliferating liver cancer cells. Cell Mol Life Sci. 2021 Jul;78(13):5397-5413. doi: 10.1007/s00018-021-03857-z.
2020
Benyelles M, O’Donohue MF, Kermasson L, Lainey E, Borie R, Lagresle-Peyrou C, Nunes H, Cazelles C, Fourrage C, Ollivier E, Marcais A, Gamez AS, Morice-Picard F, Caillaud D, Pottier N, Ménard C, Ba I, Fernandes A, Crestani B, de Villartay JP, Gleizes PE, Callebaut I, Kannengiesser C, Revy P. NHP2 deficiency impairs rRNA biogenesis and causes pulmonary fibrosis and Høyeraal-Hreidarsson syndrome. Hum Mol Genet. 2020 Apr 15;29(6):907-922. doi: 10.1093/hmg/ddaa011.
Deleye Y, Cotte AK, Hannou SA, Hennuyer N, Bernard L, Derudas B, Caron S, Legry V, Vallez E, Dorchies E, Martin N, Lancel S, Annicotte JS, Bantubungi K, Pourtier A, Raverdy V, Pattou F, Lefebvre P, Abbadie C, Staels B, Haas JT, Paumelle R. CDKN2A/p16INK4a suppresses hepatic fatty acid oxidation through the AMPKα2-SIRT1-PPARα signaling pathway. J Biol Chem. 2020 Dec 11;295(50):17310-17322. doi: 10.1074/jbc.RA120.012543.
Soquet J, Rousse N, Moussa M, Goeminne C, Deblauwe D, Vuotto F, Pontana F, Lionet A, Dubois R, Robin E, Vincentelli A. Heart retransplantation following COVID-19 illness in a heart transplant recipient. J Heart Lung Transplant. 2020 Sep;39(9):983-985. doi:
10.1016/j.healun.2020.06.026.
2019
Masclef L, Dehennaut V, Mortuaire M, Schulz C, Leturcq M, Lefebvre T, Edouart A-S. (2019) Cyclin D1 stability is partly controlled by O-GlcNAcylation. Front Endocrinol (Lausanne) 10:106.
Teissier* T, Quersin* V, Gnemmi V, Daroux M, Howsam M, Delguste F, Lemoine C, Fradin C, Schmidt AM, Cauffiez C, Brousseau T, Glowacki F, Tessier F, Boulanger E#, Frimat M#. Knock-out of receptor for advanced glycation end-products (RAGE) attenuates “physiological” age-related renal lesions. Aging Cell, 2019 Apr;18(2):e12850.
Boyer T, Gonzales F, Barthélémy A, Marceau-Renaut A, Peyrouze P, Guihard S, Lepelley P, Plesa A, Nibourel O, Delattre C, Wetterwald M, Pottier N, Plantier I, Botton S, Dombret H, Berthon C, Preudhomme C, Roumier C, Cheok M. Clinical Significance of ABCB1 in Acute Myeloid Leukemia: A Comprehensive Study. Cancers (Basel). 2019 Sep 6;11(9). pii: E1323.
Moreno Leon L, Gautier M, Allan R, Ilié M, Nottet N, Pons N, Paquet A, Lebrigand K, Truchi M, Fassy J, Magnone V, Kinnebrew G, Radovich M, Cheok MH, Barbry P, Vassaux G, Marquette CH, Ponzio G, Ivan M, Pottier N, Hofman P, Mari B, Rezzonico R. The nuclear hypoxia-regulated NLUCAT1 long non-coding RNA contributes to an aggressive phenotype in lung adenocarcinoma through regulation of oxidative stress. Oncogene. 2019 Nov;38(46):7146-7165.
2018
Sauzay C, Louandre C, Bodeau S, Anglade F, Godin C, Fontaine JX, Saidak Z, Usureau C, Molinie R, Mesnard F, Pluquet O and Galmiche A. Protein neosynthesis, a target of sorafenib, interferes with the Unfolded Protein Response (UPR) and the induction of ferroptosis in Hepatocellular carcinoma cells. Oncotarget, 2018, 9 :8400-8414
Papaioannou A, Higa A, Jégou G, Jouan F, Pineau R, Saas L, Avril T, Pluquet O and Chevet E. (2018) Alterations of EDEM1 functions enhance ATF6 pro-survival signaling. FEBS J. 285 :4146-4164
Lhomond S, Avril T, Dejeans N, McMahon M, Pineau R, Papadodima O, Voutetakis K, Logotheti M, Pallares-Lupon N, Schmit K, Le Reste PJ, Etchevery A, Mosser J, Barroso K, Vauléon E, Maurel M, Jégou G, Samali A, Patterson JB, Pluquet O, Hetz C, Quillien V, Chatziioannou A, and Chevet E. Antagonistic IRE1 RNase functions dictate glioblastoma tumor development. EMBO Mol Med. 2018, 10. Pii :e7929
2017
Xiong R, Drullion C, Verstraelen P, Demeester J, Skirtach AG, Abbadie C, De Vos WH, De Smedt SC, Braeckmans K. Fast spatial-selective delivery into live cells. J Control Release. 2017, 266:198-204
Bodeau S, Sauzay C, Pluquet O, Choukroun G and Galmiche A. A potential role of the Unfolded Protein Response in post-transplant cancer. Clin Sci (Lond). 2017, 131(13):1429-1436.
Gaudelot K, Gibier JB, Pottier N, Hémon B, Van Seuningen I, Glowacki F, Leroy X, Cauffiez C, Gnemmi V, Aubert S, Perrais M. Targeting miR-21 decreases expression of multi-drug resistant genes and promotes chemosensitivity of renal carcinoma. Tumour Biol. 2017, 39, 707372
Gibier JB, Hémon B, Fanchon M, Gaudelot K, Pottier N, Ringot B, Van Seuningen I, Glowacki F, Cauffiez C, Blum D, Copin MC, Perrais M, Gnemmi V. Dual role ofMUC1 mucin in kidney ischemia-reperfusion injury: Nephroprotector in early phase, but pro-fibrotic in late phase. Biochim Biophys Acta. 2017, 1863, 1336-1349
Nibourel O*, Guihard S*, Roumier C, Pottier N, Terre C, Paquet A, Peyrouze P, Geffroy S, Quentin S, Alberdi A, Abdelali RB, Renneville A, Demay C, Celli-LebrasK, Barbry P, Quesnel B, Castaigne S, Dombret H, Soulier J, Preudhomme C*, Cheok MH*. Copy-number analysis identified new prognostic marker in acute myeloid leukemia. Leukemia. 2017, 31, 555-564
Ghisdal L, Baron C, Lebranchu Y, Viklický O, Konarikova A, Naesens M, Kuypers D, Dinic M, Alamartine E, Touchard G, Antoine T, Essig M, Rerolle JP, Merville P, Taupin JL, Le Meur Y, Grall-Jezequel A, Glowacki F, Noël C, Legendre C, Anglicheau D, Broeders N, Coppieters W, Docampo E, Georges M, Ajarchouh Z, Massart A, Racapé J, Abramowicz D, Abramowicz M. Genome-Wide Association Study of Acute Renal Graft Rejection. Am J Transplant. 2017, 17, 201-209 Bertero T, Rezzonico R, Pottier N, Mari B. Impact of MicroRNAs in the Cellular Response to Hypoxia.
Int Rev Cell Mol Biol. 2017;333:91-158.
2016
Houessinon A, Gicquel A, Bochereau F, Louandre C, Nyga R, Godin C, Degonville J, Fournier E, Saidak Z, Drullion C, Barbare JC, Chauffert B, François C, Pluquet O, and Galmiche A. Alpha-fetoprotein is a biomarker of unfolded protein response and altered proteostasis in hepatocellular carcinoma cells exposed to sorafenib. Cancer Lett. 2016, 370(2):242-9.
Paget, M. Dubuissez, V. Dehennaut, J. Nassour, BT. Harmon, N. Spruyt, I. Loison, C. Abbadie, BR. Rood, D. Leprince. HIC1 (hypermethylated in cancer 1) SUMOylation is dispensable for DNA repair but is essential for the apoptotic DNA damage response (DDR) to irreparable DNA double-strand breaks (DSBs). Oncotarget, 2016, 8, 2916-2935
Steenackers A, Olivier-Van Stichelen S, Baldini SF, Dehennaut V, Toillon RA, Le Bourhis X, El Yazidi-Belkoura I, Lefebvre T. (2016) Silencing the Nucleocytoplasmic O-GlcNAc Transferase Reduces Proliferation, Adhesion, and Migration of Cancer and Fetal Human Colon Cell Lines. Front Endocrinol (Lausanne).25:7:46
2015
Vercoutter-Edouart AS, Yazidi-Belkoura IE, Guinez C, Baldini S, Leturcq M, Mortuaire M, Mir AM, Steenackers A, Dehennaut V, Pierce A, Lefebvre T. (2015) Detection and identification of O-GlcNAcylated proteins by proteomic approaches. Proteomics 15(5-6):1039-50
Roy S, Benz F, Vargas Cardenas D, Vucur M, Gautheron J, Schneider A,Hellerbrand C, Pottier N, Alder J, Tacke F, Trautwein C, Roderburg C, Luedde T. miR-30c and miR-193 are a part of the TGF-β-dependent regulatory network controlling extracellular matrix genes in liver fibrosis. J Dig Dis. 2015, 16, 513-524.
PUBLICATIONS CLINIQUES
2023
Hamroun A, Prouteau C, Lenain R, Roger C, Bauters A, Zawadzki C, Subtil D, Gibier JB, Stichelbout M, Coppo Paul, Lionet A, Maanaoui M, Hazzan M, Provôt F. The challenging follow‑up of pregnancy in women with known thrombotic thrombocytopenic purpura: a single‑center experience of a preemptive management protocol. Journal of Nephrology, in press
Fages V, Decaestecker A, Lessore C, Gaillard V, Odou MF, Lemaitre M, Glowacki F, Lionet A. A Pregnant Woman With Hypercalcemia-Induced Acute Pancreatitis. Kidney Int Rep. 2023 May 17;8(8):1680-1682. doi: 10.1016/j.ekir.2023.05.006.
Laboux T, Maanaoui M, Allain F, Boulanger E, Denys A, Gibier JB, Glowacki F, Grolaux G, Grunenwald A, Howsam M, Lancel S, Lebas C, Lopez B, Roumenina L, Provôt F, Gnemmi V, Frimat M. Hemolysis is associated with altered heparan sulfate of the endothelial glycocalyx and with local complement activation in thrombotic microangiopathies. Kidney Int. 2023 Aug;104(2):353-366. doi: 10.1016/j.kint.2023.03.039.
Fages V, Jannin A, Maanaoui M, Glowacki F, Do Cao C. Proteinuria reduction with SGLT2 inhibitors in a patient treated with tyrosine kinase inhibitor lenvatinib. J Nephrol. 2023 Jul 7. doi: 10.1007/s40620-023-01701-0.
Pépin M, Levassort H, Boucquemont J, Lambert O, Alencar de Pinho N, Turinici M, Helmer C, Metzger M, Cheddani L, Frimat L, Combe C, Fouque D, Laville M, Ayav C, Liabeuf S, Jacquelinet C, Teillet L, Stengel B, Massy ZA; CKD-REIN Study Collaborators. Cognitive performance is associated with glomerular filtration rate in patients with chronic kidney disease: results from the CKD-REIN cohort. J Neurol Neurosurg Psychiatry. 2023 Jan 24. PMID: 36693722.
Massart A, Danger R, Olsen C, Emond MJ, Viklicky O, Jacquemin V, Soblet J, Duerinckx S, Croes D, Perazzolo C, Hruba P, Daneels D, Caljon B, Sever MS, Pascual J, Miglinas M; Renal Tolerance Investigators; Pirson I, Ghisdal L, Smits G, Giral M, Abramowicz D, Abramowicz M, Brouard S. An exome-wide study of renal operational tolerance. Front Med (Lausanne). 2023 May 17;9:976248. doi: 10.3389/fmed.2022.976248.
Lemaitre M, Lionet A, Fages V, Vantyghem MC, Subtil D, Vambergue A. Case report: Non-PTH-dependent hypercalcemia in pregnancy: Consider CYP24A1 mutations. Ann Endocrinol (Paris). 2023 Jun 13:S0003-4266(23)00112-9. doi: 10.1016/j.ando.2023.05.009.
Bouquerel M, Dequidt L, Jullie ML, Beltzung F, Gibier JB, Lefevre G, Dezoteux F, Morice-Picard F, Trimouille A, Doutre MS. Wells syndrome and acquired cutis laxa: An atypical association. J Dermatol. 2023 Jul 12. doi: 10.1111/1346-8138.16897.
Hamdini L, Ydee A, Larsen S, Gibier JB, Azar R, Petit V. Hydroxocobalamin- induced oxalate nephropathy after smoke inhalation. J Nephrol. 2023 Jun;36(5):1443-1445. doi: 10.1007/s40620-023-01592-1.
Dezoteux F, Bongiovanni A, Tardivel M, Dendooven A, Gibier JB, Mortuaire G, Audry S, Gevaert MH, Van Poucke N, Anglo E, Lefèvre G, Staumont-Sallé D. Automatic quantification method of eosinophilic degranulation in tissues: Application for the study of eosinophilic disorders. Clin Exp Allergy. 2023 Apr 18. doi: 10.1111/cea.14323.
2022
Uhlin F, Szpirt W, Kronbichler A, Bruchfeld A, Soveri I, Rostaing L, Daugas E, Lionet A, Kamar N, Rafat C, Mysliveček M, Tesař V, Fernström A, Kjellman C, Elfving C, McAdoo S, Mölne J, Bajema I, Sonesson E, Segelmark M. Endopeptidase Cleavage of Anti-Glomerular Basement Membrane Antibodies in vivo in Severe Kidney Disease: An Open-Label Phase 2a Study. J Am Soc Nephrol. 2022 Apr;33(4):829-838. doi: 10.1681/ASN.2021111460.
Boyle EM, Baillet C, Dupré C, Lassailly G, Vuotto F, Hazzan M, Terriou L, Morschhauser F, Lionet A, Frimat M. Bartonellosis mimicking post-transplant lymphoproliferative diseases. Nephrol Dial Transplant. 2022 Feb 25;37(3):599-601.
Hamroun A, Lenain R, Gibier JB, Maanaoui M, Lionet A. Nephrology picture: intraglomerular metastases, an exceptional cause of glomerulonephritis. J Nephrol. 2022 Jan;35(1):361-362. doi: 10.1007/s40620-021-00999-y.
Hamroun A, Speyer E, Ayav C, Combe C, Fouque D, Jacquelinet C, Laville M, Liabeuf S, Massy ZA, Pecoits-Filho R, Robinson BM, Glowacki F, Stengel B, Frimat L; CKD-REIN study Collaborators. Barriers to conservative care from patients’ and nephrologists’ perspectives: the CKD-REIN study. Nephrol Dial Transplant. 2022 Nov 23;37(12):2438-2448. doi: 10.1093/ndt/gfac009.
Hamroun A, Frimat L, Laville M, Metzger M, Combe C, Fouque D, Jacquelinet C, Ayav C, Liabeuf S, Lange C, Herpe YE, Zee J, Glowacki F, Massy ZA, Robinson B, Stengel B; Chronic Kidney Disease-Renal Epidemiology and Information Network (CKD-REIN) study group. New insights into acute-on-chronic kidney disease in nephrology patients: the CKD-REIN study. Nephrol Dial Transplant. 2022 Aug 22;37(9):1700-1709. doi: 10.1093/ndt/gfab249..
Villain C, Metzger M, Liabeuf S, Hamroun A, Laville S, Mansencal N, Combe C, Fouque D, Frimat L, Jacquelinet C, Laville M, Ayav C, Briançon S, Pecoits-Filho R, Hannedouche T, Stengel B, Massy ZA; CKD-REIN Study Group. Effectiveness and Tolerance of Renin-Angiotensin System Inhibitors With Aging in Chronic Kidney Disease. J Am Med Dir Assoc. 2022 Jun;23(6):998-1004.e7. doi: 10.1016/j.jamda.2021.10.019.
El Karoui K, Hourmant M, Ayav C, Glowacki F, Couchoud C, Lapidus N; REIN Registry. Vaccination and COVID-19 Dynamics in Dialysis Patients. Clin J Am Soc Nephrol. 2022 Mar;17(3):395-402. doi: 10.2215/CJN.10300721.
Laville SM, Couturier A, Lambert O, Metzger M, Mansencal N, Jacquelinet C, Laville M, Frimat L, Fouque D, Combe C, Robinson BM, Stengel B, Liabeuf S, Massy ZA; CKD-REIN study collaborators. Urea levels and cardiovascular disease in patients with chronic kidney disease. Nephrol Dial Transplant. 2022 Feb 26;38(1):184–92. doi: 10.1093/ndt/gfac045.
Zaworski J, Gnemmi V, Bataille P, Hachulla E, Glowacki F, Gibier JB, Daroux M, Ratsimbazafy A, Bitton L, Humez S, Guincestre T, Béhal H, Azar R, Hoffmann M, Cardon G, Bourdon F, Lemoine C, Auxenfant E, Copin MC, Vandenbussche C, Quéméneur T; pour la cohorte RENVAS. Early Renal Recovery after the First Flare of Pauci-Immune Glomerulonephritis. Am J Nephrol. 2022;53(1):59-68. doi: 10.1159/000520285.
Colombat M, Gaspard M, Camus M, Dalloux-Chioccioli J, Delas A, Poullot E, Moktefi A, François A, Moreau A, Gibier JB, Raynaud P, Huart A, Piedrafita A, Gilhodes J, Lairez O, Grateau G, Georgin-Lavialle S, Maisonneuve H, Moreau P, Jaccard A, Bridoux F, Plante-Bordeneuve V, Damy T, Mal H, Brousset P, Valleix S, Burlet-Schiltz O. Mass spectrometry-based proteomics in clinical practice amyloid typing: state-of-the-art from a French nationwide cohort. Haematologica. 2022 Dec 1;107(12):2983-2987. doi: 10.3324/haematol.2022.281431.
Fourdinier O, Ulrich M, Karras A, Olagne J, Buob D, Audard V, Vigneau C, Gibier JB, Guerrot D, Massy Z, Vuiblet V, Rabot N, Goujon JM, Cordonnier C, Choukroun G, Titeca-Beauport D. Glomerulonephritis with non-Randall-type, non- cryoglobulinaemic monoclonal immunoglobulin G deposits (PGNMID and ITG). Clin Kidney J. 2022 Mar 24;15(9):1727-1736. doi: 10.1093/ckj/sfac085.
Martins M, Bridoux F, Goujon JM, Meuleman MS, Ribes D, Rondeau E, Guerry MJ, Delmas Y, Levy B, Ducloux D, Kandel-Aznar C, Le Fur A, Garrouste C, Provot F, Gibier JB, Thervet E, Bruneval P, Rabant M, Karras A, Dragon Durey MA, Fremeaux- Bacchi V, Chauvet S. Complement Activation and Thrombotic Microangiopathy Associated With Monoclonal Gammopathy: A National French Case Series. Am J Kidney Dis. 2022 Sep;80(3):341-352. doi: 10.1053/j.ajkd.2021.12.014.
Chastagner M, Shourik J, Jachiet M, Battistella M, Lefevre G, Gibier JB, Aubert H, Musquer M, Descamps V, Deschamps L, Chosidow O, Ortonne N, Groh M, Bernier M, Jullien D, Chasset F, Staumont-Salle D, Bouaziz JD, Kanitakis J, Villani AP. Treatment of Eosinophilic Annular Erythema: Retrospective multicenter study and literature review. Ann Dermatol Venereol. 2022 Jun;149(2):123-127. doi: 10.1016/j.annder.2021.07.007.
Lemarchant B, Lebouvier T, Delbeuck X, Gibier JB, Tard C. A case of transthyretin-related cerebral amyloid angiopathy. The other side of hereditary transthyretin amyloidosis. Acta Neurol Belg. 2022 Apr;122(2):571-573. doi: 10.1007/s13760-021-01854-4.
2021
Laboux T, Gibier JB, Pottier N, Glowacki F, Hamroun A. COVID-19-related collapsing glomerulopathy revealing a rare risk variant of APOL1: lessons for the clinical nephrologist. J Nephrol. 2021 Feb 6:1–6. doi: 10.1007/s40620-020-00935-6.
Epub ahead of print. PMID: 33548053; PMCID: PMC7865108.
Hamroun A, Camier A, Bigna JJ, Glowacki F. Impact of air pollution on renal outcomes: a systematic review and meta-analysis protocol. BMJ Open. 2021 Jan 17;11(1):e041088. doi: 10.1136/bmjopen-2020-041088. PMID: 33455930; PMCID: PMC7813312.
2020
Gnemmi V, Gibier JB, Humez S, Copin MC, Glowacki F. Néphrite interstitielle granulomateuse : le point de vue du pathologiste [Renal granulomatous nephritis: Histopathological point of view]. Ann Pathol. 2020 Dec 1:S0242-6498(20)30267-4. French. doi: 10.1016/j.annpat.2020.11.001. Epub ahead of print. PMID: 33277052.
Couchoud C, Bayer F, Ayav C, Béchade C, Brunet P, Chantrel F, Frimat L, Galland R, Hourmant M, Laurain E, Lobbedez T, Mercadal L, Moranne O; French REIN registry. Low incidence of SARS-CoV-2, risk factors of mortality and the course of illness in the French national cohort of dialysis patients. Kidney Int. 2020 Dec;98(6):1519-1529. doi: 10.1016/j.kint.2020.07.042. Epub 2020 Aug 25. PMID: 32858081; PMCID: PMC7445552.
Alencar de Pinho N, Kaboré J, Laville M, Metzger M, Lange C, Jacquelinet C, Combe C, Fouque D, Frimat L, Ayav C, Robinson BM, Drueke T, Massy ZA, Stengel B; CKD-REIN Study Group. Urinary Sodium-to-Potassium Ratio and Blood Pressure in CKD. Kidney Int Rep. 2020 Jun 2;5(8):1240-1250. doi: 10.1016/j.ekir.2020.05.025. PMID: 32775823; PMCID: PMC7403539.
Paccou J, Pflimlin A, Glowacki F, Cortet B. A Challenging Case of Tumor-Induced Osteomalacia. Am J Med. 2021 Jan;134(1):e60-e61. doi: 10.1016/j.amjmed.2020.06.032. Epub 2020 Jul 24. PMID: 32712146.
Petit V, Bonnafous P, Fages V, Gautheret-Dejean A, Engelmann I, Baras A, Hober D, Gérard R, Gibier JB, Leteurtre E, Glowacki F, Moulonguet F, Decaestecker A, Provôt F, Chamley P, Faure E, Prusty BK, Maanaoui M, Hazzan M. Donor-to-recipient transmission and reactivation in a kidney transplant recipient of an inherited chromosomally integrated HHV-6A: Evidence and outcomes. Am J Transplant. 2020 Dec;20(12):3667-3672. doi: 10.1111/ajt.16067. Epub 2020 Jun 28. PMID: 32428994.
Perry A, Douillard C, Jonca F, Glowacki F, Leroy X, Caveriviere P, Hubert A, Labrune P. Papillary renal cell carcinoma in two young adults with glycogen storage disease type Ia. JIMD Rep. 2020 Jan 29;52(1):17-22. doi: 10.1002/jmd2.12096. PMID: 32154055; PMCID: PMC7052693.
Delsart P, Vambergue A, Ninni S, Machuron F, Lelievre B, Ledieu G, Fontaine P, Merlen E, Frimat M, Glowacki F, Montaigne D, Mounier-Vehier C. Prognostic significance of the renal resistive index in the primary prevention of type II diabetes. J Clin Hypertens (Greenwich). 2020 Feb;22(2):223-230. doi: 10.1111/jch.13819. Epub 2020 Jan 31. PMID: 32003935.
Bitton L, Vandenbussche C, Wayolle N, Gibier JB, Cordonnier C, Verine J, Humez S, Bataille P, Lenain R, Ramdane N, Azar R, Mac Namara E, Hatron PY, Maurage CA, Perrais M, Frimat M, Vanhille P, Glowacki F, Buob D, Copin MC, Quéméneur T, Gnemmi V. Tubulointerstitial damage and interstitial immune cell phenotypes are useful predictors for renal survival and relapse in antineutrophil cytoplasmic antibody-associated vasculitis. J Nephrol. 2020 Aug;33(4):771-781. doi: 10.1007/s40620-019-00695-y. Epub 2020 Jan 8. PMID: 31916228.
2019
Robert L, Ficheur G, Gautier S, Servais A, Luyckx M, Soula J, Decaudin B,Glowacki F, Puisieux F, Chazard E, Beuscart JB. Community-Acquired Acute Kidney Injury Induced By Drugs In Older Patients: A Multifactorial Event. Clin IntervAging. 2019 Dec 5;14:2105-2113.
Hamroun A, Frimat M, Beuscart JB, Buob D, Lionet A, Lebas C, Daroux M, Provôt F, Hazzan M, Boulanger É, Glowacki F. [Kidney disease care for the elderly].Nephrol Ther. 2019 Dec;15(7):533-552.
Pekar JD, Grzych G, Durand G, Haas J, Lionet A, Brousseau T, Glowacki F, Maboudou P. Calcium state estimation by total calcium: the evidence to end thenever-ending story. Clin Chem Lab Med. 2019 Sep 2. pii:/j/cclm.ahead-of-print/cclm-2019-0568/cclm-2019-0568.xml.
Vandenbussche C, Bitton L, Bataille P, Glowacki F, Azar R, Hatron PY,Macnamara E, Gheerbrant JD, Cardon G, Hoffmann M, Auxenfants E, Gnemmi V,Quéméneur T. Prognostic Value of Microscopic Hematuria after Induction ofRemission in Antineutrophil Cytoplasmic Antibodies-Associated Vasculitis. Am J Nephrol. 2019;49(6):479-486.
Artru F, Louvet A, Glowacki F, Bellati S, Frimat M, Gomis S, Castel H,Barthelon J, Lassailly G, Dharancy S, Noel C, Hazzan M, Mathurin P. Theprognostic impact of cirrhosis on patients receiving maintenance haemodialysis. Aliment Pharmacol Ther. 2019 Jul;50(1):75-83.
Vigneau C, Ayav C, Noël N, Gomis S, Glaudet F, Siébert M, Kessler M, Nogier MB, Villar E, Allot V, Edet S, Glowacki F, Baudoin V, Allain-Launay E, Dunand O, Moranne O, Hogan J, Couchoud C; registre REIN. [Towards an extension of the REIN registry to patients with chronic kidney disease at stage 5 not treated withdialysis or transplantation? A pilot study]. Nephrol Ther. 2019Jun;15(3):143-151.
2018
Duployez N, Marceau-Renaut A, Villenet C, Petit A, Rousseau A, Ng SWK, Paquet A, Gonzales F, Barthélémy A, Leprêtre F, Pottier N, Nelken B, Michel G, Baruchel A, Bertrand Y, Leverger G, Lapillonne H, Figeac M, Dick JE, Wang JCY, Preudhomme C, Cheok M. The stem cell-associated gene expression signature allows risk stratification in pediatric acute myeloid leukemia. Leukemia. 2018
Zaworski J, Frimat M, Duval M, Saint-Jacques C, Bouderlique É, Glowacki F, Noël C, Hazzan M, Provôt F. [Vancomycin poisoning successfully treated with intermittent hemodialysis: A case report]. Nephrol Ther. 2018, 14, 112-116
Châtelet V, Lobbedez T, Harambat J, Bayat-Makoei S, Glowacki F, Vigneau C. [Socioeconomic inequalities and kidney transplantation]. Nephrol Ther. 2018, 14, 81-84
2017
Rabant M, Boullenger F, Gnemmi V, Pellé G, Glowacki F, Hertig A, Brocheriou I, Suberbielle C, Taupin JL, Anglicheau D, Legendre C, Duong Van Huyen JP, Buob D. Isolated v-lesion in kidney transplant recipients: Characteristics, association with DSA, and histological follow-up. Am J Transplant. 2017, 18, 972-981
Gibier JB, Gnemmi V, Glowacki F, Boyle EM, Lopez B, MacNamara E, Hoffmann M,Azar R, Guincestre T, Bourdon F, Copin MC, Buob D. Intratubular amyloid in light chain cast nephropathy is a risk factor for systemic light chain amyloidosis. Mod Pathol. 2017, 31, 452-462
Belaiche S, Mercier E, Cuny D, Kambia N, Wierre P, Bertoux É, Mascaut D, Azar R, Bataille P, Bourdon F, Mac Namara É, Maisonneuve N, Painchart B, Vrigneau L,Noël C, Décaudin B, Glowacki F; réseau Néphronor. [Community pharmacists’interventions to prevent and screen chronic kidney disease patients]. Nephrol Ther. 2017, 13, 87-92
2016
Tabibzadeh N, Glowacki F, Frimat M, Elsermans V, Provôt F, Lionet A, Gnemmi V, Hertig A, Noël C, Hazzan M. Long-term outcome after early cyclosporine withdrawal in kidney transplantation: ten years after. Clin Transplant. 2016, 30, 1480-1487
Buob D, Decambron M, Gnemmi V, Frimat M, Hoffmann M, Azar R, Gheerbrant JD, Guincestre T, Noël C, Copin MC, Glowacki F. Collapsing glomerulopathy is common in the setting of thrombotic microangiopathy of the native kidney. Kidney Int. 2016, 90, 1321-1331
Hannedouche T, Roth H, Krummel T, London GM, Jean G, Bouchet JL, Drüeke TB, Fouque D; French Observatory. Multiphasic effects of blood pressure on survival in hemodialysis patients. Kidney Int. 2016, 90, 674-684
Legendre M, Devilliers H, Perard L, Groh M, Nefti H, Dussol B, Trad S, Touré F, Abad S, Boffa JJ, Frimat L, Torner S, Seidowsky A, Massy ZA, Saadoun D, RieuV, Schoindre Y, Heron E, Frouget T, Lionet A, Glowacki F, Arnaud L, Mousson C,Besancenot JF, Rebibou JM, Bielefeld P. Clinicopathologic characteristics,treatment, and outcomes of tubulointerstitial nephritis and uveitis syndrome in adults: A national retrospective strobe-compliant study. Medicine (Baltimore).2016, 95, e3964
Frimat M*, Decambron M*, Lebas C, Moktefi A, Lemaitre L, Gnemmi V, Sautenet B, Glowacki F, Subtil D, Jourdain M, Rigouzzo A, Brocheriou I, Halimi JM, Rondeau E,Noel C, Provôt F, Hertig A. Renal Cortical Necrosis in Postpartum Hemorrhage: A Case Series. Am J Kidney Dis. 2016, 68, 50-57
Massart A*, Pallier A*, Pascual J, Viklicky O, Budde K, Spasovski G, Klinger M,Sever MS, Sørensen SS, Hadaya K, Oberbauer R, Dudley C, De Fijter JW, Yussim A,Hazzan M, Wekerle T, Berglund D, De Biase C, Pérez-Sáez MJ, Mühlfeld A, OrlandoG, Clemente K, Lai Q, Pisani F, Kandus A, Baas M, Bemelman F, Ponikvar JB, MazouzH, Stratta P, Subra JF, Villemain F, Hoitsma A, Braun L, Cantarell MC, Colak H,Courtney A, Frasca GM, Howse M, Naesens M, Reischig T, Serón D, Seyahi N, Tugmen C, Alonso Hernandez A, Beňa L, Biancone L, Cuna V, Díaz-Corte C, Dufay A,Gaasbeek A, Garnier A, Gatault P, Gentil Govantes MA, Glowacki F, Gross O,Hurault de Ligny B, Huynh-Do U, Janbon B, Jiménez Del Cerro LA, Keller F, LaManna G, Lauzurica R, Le Monies De Sagazan H, Thaiss F, Legendre C, Martin S,Moal MC, Noël C, Pillebout E, Piredda GB, Puga AR, Sulowicz W, Tuglular S,Prokopova M, Chesneau M, Le Moine A, Guérif P, Soulillou JP, Abramowicz M, Giral M, Racapé J, Maggiore U, Brouard S*, Abramowicz D*. The DESCARTES-Nantes survey of kidney transplant recipients displaying clinical operational tolerance identifies35 new tolerant patients and 34 almost tolerant patients. Nephrol Dial Transplant. 2016, 31, 1002-1013
2015
Diss M, Ranchin B, Broly F, Pottier N, Cochat P. [Type 1 xanthinuria: Report on three cases]. Arch Pediatr. 2015, 22, 1288-1291
Jamet MP, Gnemmi V, Hachulla É, Dhaenens CM, Bouchindhomme B, Delattre C, Glowacki F, Hatron PY, Lacour A, Lamblin N, Launay D, Leleu X, Guiochon-Mantel A,Valleix S, Maurage CA, Copin MC, Buob D. Distinctive Patterns of Transthyretin Amyloid in Salivary Tissue: A Clinicopathologic Study of 92 Patients With Amyloid-containing Minor Salivary Gland Biopsies. Am J Surg Pathol. 2015, 39, 1035-1044
Gnemmi V, Verine J, Vrigneaud L, Glowacki F, Ratsimbazafy A, Copin MC, Dewilde A, Buob D. Microvascular inflammation and acute tubular necrosis are major histologic features of hantavirus nephropathy. Hum Pathol. 2015, 46, 827-835
Thèses en cours
- Fellah Sandy (D4 en 2023 – Directeur Thèse: Christelle Cauffiez)
- Joëlle Giroud (D3 en 2023 – Directeur Thèse: Olivier Pluquet)
- Romain Larrue (D3 en 2023 – Directeur Thèse: Nicolas Pottier)
- Henry Abi Rached (D2 en 2023 – Directeur Thèse: Corinne Abbadie/Laurent Mortier)
- Elodie Rozinski (D2 en 2023 – Directeur Thèse: Corinne Abbadie)
HDR soutenues
- Dr Vanessa Dehennaut (soutenue le 16 juillet 2020 à l’université de Lille) sur le thème « La O-GlcNAcylation: un biomarqueur et une cible thérapeutique potentielle des cancers colorectaux » (section CNU64)
- Dr Cynthia Van der Hauwaert (soutenue le 23 juin 2021 à l’université de Lille) sur le thème « Identification et caractérisation d’ARN non-codants dans les processus de fibrose rénale et à l’origine de la résistance des tumeurs pulmonaires aux anticancéreux » (section CNU4400)
Thèses soutenues
- LEMAIRE Julie (soutenue le 25/01/2021 dirigée par : Nicolas Pottier
- DECOURCELLE Amélie (soutenue le 11/12/2020) dirigée par : Vanessa Dehennaut
- ABDELFETTAH Souhila (soutenue le 19/12/2019) dirigée par : Dominique LEPRINCE
- DEWAELES Edmone (soutenue le 05/11/2019) dirigée par : Christelle CAUFFIEZ and David BLUM
- TEISSIER Thibault (soutenue le 20/09/2019) dirigée par : Christelle CAUFFIEZ and Eric BOULANGER
- HENNINO Marie-Flore (soutenue le 28/04/2017) dirigée par: François GLOWACKI and Christelle CAUFFIEZ
- SAVARY Grégoire (soutenue le 20/12/2016) dirigée par: Christelle CAUFFIEZ and Bernard MARI
- PAGET Sonia (soutenue le 15/12/2016) dirigée par: Dominique LEPRINCE
- TOMEZAK Maxime (soutenue le xx/12/2016) dirigée par : Fabrizio CLERI and Corinne ABBADIE
- DUBUISSEZ Marion (soutenue le 05/10/2015) dirigée par : Dominique LEPRINCE
- NASSOUR Joe (soutenue le xx/09/2015) dirigée par: Corinne ABBADIE
Contenu de l’onglet